
四面体型中間体
四面体型中間体(しめんたいがたちゅうかんたい)もしくは四面体中間体(英語: Tetrahedral intermediate、TI[1])は炭素原子の周りで結合の組み換えが起こり、二重結合を持つ平面三角形の炭素が四面体型のsp3炭素に変わるときに生成する反応中間体である[2]。四面体型中間体はカルボニル基への求核付加によって炭素-酸素結合のπ結合が切れて生成する[3]。四面体型中間体の安定性は新しいsp3炭素に結合している、負電荷を持った脱離基の脱離能に依存する。もし元から結合していた基と新しくカルボニルに結合する基の両方が電気的に陰性である場合、四面体型中間体は不安定である[3]。四面体型中間体はエステル化やエステル交換反応、エステルの加水分解、アミドやペプチドの合成や加水分解、ヒドリド還元などの反応で鍵となる中間体であるため、これらの反応を扱う有機合成や生物学系できわめて重要である。
歴史
四面体中間体が最初に言及されたのは1887年、ルートヴィヒ・クライゼンの論文の中である[4]。安息香酸ベンジルとナトリウムメトキシド、および安息香酸メチルとナトリウムベンジルオキシドの反応で、彼は酸性条件下で安息香酸ベンジル、安息香酸メチル、メタノール、ベンジルアルコールを生成する白色沈殿を発見した。彼はこれらに共通の中間体を"additionelle Verbindung"(付加化合物)と命名した。[要出典]

ヴィクトル・グリニャールは1901年、エステルとグリニャール試薬の反応を調べる中で、反応中に不安定な中間体が生成していると考えた[5]。
カルボン酸誘導体の置換反応中に四面体中間体が生成しているということの根拠を最初に示したのはマイロン・リー・ベンダーで、1951年のことであった[6]。彼はカルボン酸誘導体のカルボニル酸素を同位体である18O(英語版)で同位体標識(英語版)して水と反応させ、ラベルされたカルボン酸をつくった[6]。彼が反応のあとに残っていた原料を調べたところ、ラベルされた酸素を含む原料の割合が減っていることに気づいた。これは四面体型中間体の生成を示している[6]。
反応機構

カルボニル基への求核攻撃はビュルギ・ダニッツ軌跡を通って進行する。求核剤の電子軌道とC-O結合の軌道のなす角度は90˚より大きい。この角度で、求核剤のHOMO(最高被占軌道)とC-O二重結合のπ*LUMO(最低空軌道)がもっとも軌道の重なりが大きくなる。[要出典]
四面体型中間体が分解する際は、より弱い塩基が容易に脱離する[1]。強塩基が結合したカルボニル化合物に弱塩基が近づいてきて四面体型中間体が生成しても、より優れた脱離基である弱塩基(あとから入ってきたほう)が先に抜けてしまうので、反応は進行しない[1]。脱離基が弱塩基であるときは系の自由エネルギーが小さく、強塩基であるときは大きい[1]。反応は自由エネルギーが小さくなる方向に進む[1]。
カルボニル炭素に電気的に陰性でない基(脱離すると強塩基となる基)が結合している場合、四面体型中間体は安定である[7]。この場合、求核剤が近づくと不可逆的な求核付加反応がおこる[7]。
エステルの加水分解では途中で四面体型中間体が生成するが、素反応のうち四面体型中間体ができる反応が律速段階(最も遅い反応)、四面体型中間体から塩基が脱離する反応が2番目に遅い反応になっている[8]。エステル加水分解で用いられる酸触媒は、カルボニル酸素をプロトン化することでプロトン化されていないカルボニル酸素よりも求核付加を受けやすくし、四面体型中間体の生成速度を増大させる[8]。また四面体型中間体からアルコールが抜ける際、酸触媒によって脱離基の塩基性が弱められるので、この反応の反応速度も大きくなる[8]。
エステルの加水分解は、四面体型中間体の生成と分解という二つの段階に大きく分けることができる[9]。
四面体型中間体の構造
一般的な特徴
四面体中間体は一時的に生成し、不安定なので[3]すぐに変化してしまうものが多いが、この形をもつ多くの化合物が知られている。[要出典]アルデヒドやケトンおよびそれらの誘導体は、グリニャール試薬やヒドリドイオン(H-)が付加する際に安定で観測可能な四面体型中間体をつくることがよくあるが[10]カルボン酸誘導体の場合はそうではない[3]。カルボン酸誘導体と同レベルまで酸化されている炭素に結合している置換基、すなわちORやOAr(エーテル)、NR2やClなどはカルボニル基と共役しているため、同じカルボニル構造をもつアルデヒドやケトンに比べカルボニル基への求核付加が熱力学的に不利になる。すなわちこれらの基を含む四面体型中間体は不安定である[3]。 安定なカルボン酸誘導体の四面体型中間体は、以下の4つのうち少なくとも一つの特徴を持っている。[要出典]
- 多環構造(例:テトロドトキシン)[11]
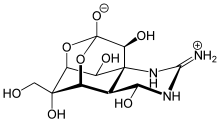
テトロドトキシン - 強い電子求引性基がアシル炭素に結合している化合物 (例:N,N-ジメチルトリフルオロアセトアミド)[12]
- カルボニル基と共役しにくい電子供与性基を含む化合物(例: シクロール)[13]
- 硫黄原子がアノマー中心に結合している化合物(例: S-アシル化-1,8-ナフタレンジチオール)[14]

これらの化合物は四面体型中間体がカルボニル化合物に分解する反応における速度論を研究するために、中間体のIRやUV、NMRの吸収スペクトルを取るために用いられる[要出典]。
X線結晶構造解析
四面体型中間体の最初のX線結晶構造解析は、1974年にイノシシのトリプシンをダイズトリプシン阻害剤とともに結晶化させて行われた[要出典]。またウシのトリプシンとアプロチニンを一緒に結晶化して構造を調べる実験は[15][16]。どちらのケースでも、四面体型中間体は酵素の活性部位に取り込まれて安定化し、ペプチド加水分解の遷移状態が安定化している[要出典]。
四面体型中間体の構造に関する考察が、1967年に結晶化されたN-ブロシルミトマイシンAの構造から得られている[17]。四面体炭素C17はO3と136.54pmの結合を形成している。これはC8-O3結合(142.31pm)より短い。対照的に、C17-N2結合(149.06pm)はN1-C1結合(148.75pm)やN1-C11結合(147.85pm)より長い。これはO3の孤立電子対がC17-N2結合のσ*軌道と相互作用するからである。しかし、このモデルは四環構造を含んでおり、カルボニル基に含まれるはずのO3がメチル化されているため、全体的にはあまりよいモデルではない。[要出典]

のちに行われた1-アザ-3,5,7-トリメチルアダマンタン-2-オンのX線結晶構造解析で、カチオン性四面体型中間体のよりよいモデルが得られた[18]。C1-N1結合は以前のデータより長い[155.2(4)pm]、C1-O1(2)結合は以前より短い[138.2(4)pm]であるとわかった[要出典]。

2002年、デイビッド・エバンズらは非常に安定で電気的に中性である四面体型中間体を、N-アシルピロールと有機金属化合物を反応させ、その後に塩化アンモニウムでプロトン化してカルビノールを得る反応のなかで観測した[19]。この物質のC1-N1結合[147.84(14) pm]は通常のCsp3-Npyrrole結合より長く、141.2-145.8 pmである。対照的に、C1-O1結合[141.15(13) pm]はCsp3-OH結合の平均的な長さ(約143.2 pm)より短い。C1-N1結合が長くなり、C1-O1結合が短くなったことは、酸素の孤立電子対とC-N結合のσ*軌道の相互作用から生じるアノマー効果で説明できる。同様に、酸素の孤立電子対とC-C結合のσ*軌道の相互作用が、C1-C2結合([152.75(15) pm]が平均的なCsp2-Csp2結合(151.3 pm)より長くなっていることの理由になっているはずである。また、C1-C11結合([152.16(17) pm])はCsp3-Csp3結合の平均(約153.0 pm)よりもやや短い。[要出典]

四面体型中間体の安定性
アセタールとヘミアセタール
ヘミアセタールとアセタールは実質的に、電気的に中性な四面体型中間体である[20]。それらは求核剤がカルボニル基に付加するときに生成するが[20]、四面体型中間体と異なり非常に安定で、有機合成化学で保護基として用いられる[21]。アセトアルデヒドがメタノールに溶解するとヘミアセタールが生成する[20]。ほとんどのヘミアセタールはもとになるアルコールやアルデヒドに比べ不安定である。例えば、アセトアルデヒドと単純なアルコールが反応してヘミアセタールができる反応の平衡定数はだいたい0.5である。[要出典]ただし平衡定数KはK=[ヘミアセタール]/[アルデヒド][アルコール]で定義される。ケトンのヘミアセタール(ヘミケタール[20])はアルデヒドのヘミアセタールよりも不安定である。しかし、環状ヘミアセタールや電子吸引基をもっているヘミアセタールは安定である。カルボニル炭素に結合した電子吸引基は平衡をヘミアセタールの側に傾ける。それらはカルボニル基の極性を増加させ、すでに正の部分電荷をもっている炭素のδ+性を増加させ、求核剤が攻撃しやすいようにする。下の図はカルボニル化合物の水和の傾向を示したものである。ヘキサフルオロアセトンは最も水和しやすいカルボニル化合物であると見られている。[要出典]ホルムアルデヒドはカルボニル基の両側が水素原子のみで立体障害がほとんどないため、水と速やかに反応する[22][23]。

シクロプロパノン(三員環の環状ケトン)も非常に水和しやすい。 三員環は環ひずみが非常に大きく(結合角が60˚まで捻じ曲げられている)、sp2混成よりはsp3混成の方が望ましい。sp2混成のケトンの結合角が約60°であるのに対し、sp3混成の水和物は結合が約49˚まで歪められている。したがってカルボニル基への付加により環構造特有のひずみが解放されるので、シクロプロパノンやシクロブタノンがとても求電子性が高い。より大きな環になると、結合角がゆがめられていないので、ヘミアセタールの安定性はエントロピーや近接性のため、小さな環に比べ高くなる。1分子のヘミアセタールが生成するのにカルボニルが2分子必要であるため、鎖式アセタールの生成ではエントロピーが減少する。対照的に、環状ヘミアセタールでは一つの分子が分子内で反応を起こすため、反応は起こりやすい。環状ヘミアセタールの安定性は平衡定数(正反応と逆反応の速度定数の比)からも理解できる。環状ヘミアセタールでは分子内反応が起こるため、求核剤は常にカルボニル基の近くにあって攻撃しやすく、正反応の速度定数が逆反応のそれに比べてずっと大きい。[要出典]グルコースなど、糖に関する生体分子はの多くは環状ヘミアセタールである[24]。

酸の存在下で、ヘミアセタールは脱離反応を起こし、もともとのアルデヒドのカルボニル基に含まれていた酸素原子が水分子として抜ける[20]。これらのオキソニウムイオンは強力な求電子剤で、2分子目と速やかに反応してより安定な化合物、アセタールを与える[20]。ヘミアセタールからアセタールが生成する反応機構は以下の通り[20]。

アセタールは、上述のとおり安定な四面体型中間体であるため、有機合成化学において保護基として用いられる[21]。塩基性条件下ではアセタールは安定で、塩基からケトンを保護するのに用いられる[21]。アセタール基は酸性条件下で加水分解され、アルデヒドやケトンへと戻る[25]。ジオキソランを保護基として用いる場合の例を下に示す。

ワインレブアミド
ワインレブアミドは、N-メトキシ-N-メチルカルボン酸アミドのことである[26]。ワインレブアミドは有機金属化合物と反応してプロトン化されたケトンを与える(ワインレブのケトン合成)。五員環キレート中間体が高い安定性を示すため、ケトンの収率が高く、この方法は広く受け入れられている。[要出典]量子化学計算により安定な四面体型付加体ができやすいことがわかっており、これは実験結果と一致する[27]。ワインレブアミドとアルキルリチウムやグリニャール試薬との反応性の高さは四面体型付加体のキレートによる安定化、もっといえば付加体に至る遷移状態の安定性に由来している[要出典]。四面体付加の反応式を以下に示す。

医学への応用
薬の設計
タンパク質に結合している溶媒和した配位子はいくつかの立体配座が混じった平衡混合物になっていると考えられている。溶媒和したタンパク質も同様にいくつかの配座の平衡混合物になっている。タンパク質と配位子の錯体形成には、配位結合の位置を決める溶媒分子の置換も含まれており、溶媒和錯体が形成される。これがエントロピー的に好ましくないため、エンタルピー的に理想的なタンパク質と配位子の接触では、エントロピーは小さくならざるを得ない。標的のタンパク質に関する新しい配位子のデザインは、すでに知られている配位子の修正をベースに設計される。[要出典]プロテアーゼはペプチド結合の加水分解を触媒する酵素である[28]。これらのタンパク質はペプチドの加水分解反応で遷移状態にある四面体型中間体を認識し、結合するように進化してきた。ゆえに、主なプロテアーゼ阻害剤は四面体型中間体に擬態し、アルコールやリン酸基をもつ物質である。[要出典]これらの阻害剤の例としてサキナビル、リトナビル、ペプスタチンなどが挙げられる[29]。
酵素活性
酵素の活性部位内での四面体型中間体の安定化の研究は、四面体型中間体に擬態する化合物を使って調査されてきた。遷移状態の安定化に関わる結合の力は結晶学的に記述されてきた。[要出典]哺乳綱のセリンプロテアーゼ、トリプシンやキモトリプシン、ポリペプチド骨格のうち2つのN-H結合からできる「オキシアニオンホール」が負の部分電荷をもつ酸素原子と四面体型中間体の水素結合に寄与している[30]。簡単な相互作用の図を下に示す。

脚注
- ^ a b c d e 「ブルース有機化学 第7版 下」p.826
- ^ “IUPAC Gold Book definition”. 2018年1月29日閲覧。
- ^ a b c d e 「ブルース有機化学 第7版 下」p.822
- ^ Claisen, L. (1887). “Ueber die Einwirkung von Natriumalkylaten auf Benzaldehyd”. Chemische Berichte 20 (1): 646–650. doi:10.1002/cber.188702001148.
- ^ Grignard, V. (1901). “Mixed organomagnesium combinations and their application in acid, alcohol, and hydrocarbon synthesis”. Ann. Chim. Phys. 24: 433–490.
- ^ a b c Bender, M. L. (1951). “Oxygen Exchange as Evidence for the Existence of an Intermediate in Ester Hydrolysis”. アメリカ化学会誌 73 (4): 1626–1629. doi:10.1021/ja01148a063.
- ^ a b 「ブルース有機化学 第7版 下」p.891
- ^ a b c 「ブルース有機化学 第7版 下」p.1238
- ^ 「ブルース有機化学 第7版 下」p.1239
- ^ 「ブルース有機化学 第7版 下」p.910
- ^ Woodward, R. B.; Gougoutas, J. Z. (1964). “The Structure of Tetrodotoxin”. 米国化学会誌 86 (22): 5030. doi:10.1021/ja01076a076.
- ^ Gideon, Fraenkel; Watson Debra (1975). “Alkoxide adduct of an amide. Mean lifetime of an intimate ion pair”. 米国化学会誌 97 (1): 231–232. doi:10.1021/ja00834a063.
- ^ Cerrini, S.; Fedeli W.; Mazza F. (1971). “X-Ray crystallographic proof of a cyclol structure in a tripeptide”. Chemical Communications (24): 1607–1608. doi:10.1039/C29710001607.
- ^ Tagaki, M.; Ishahara R.; Matsudu T. (1977). “Mono S-Acylated 1,8-Naphthalenedithiol. Isolation and Characterization of Tetrahedral Intermediate in the Intramolecular Acyl Transfer Reaction”. 日本化学会欧文誌 50 (8): 2193–2194. doi:10.1246/bcsj.50.2193.
- ^ Sweet, R.M.; Wright H.T.; Clothia C.H.; Blow D.M. (1974). “Crystal structure of the complex of porcine trypsin with soybean trypsin inhibitor (Kunitz) at 2.6 Å resolution”. Biochemistry 13 (20): 4212–4228. doi:10.1021/bi00717a024. PMID 4472048.
- ^ Ruhlmann, A.; Kukla D.; Schwager P.; Bartels K.; Huber R. (1973). “Structure of the complex formed by bovine trypsin and bovine pancreatic trypsin inhibitor. Crystal structure determination and stereochemistry of the contact region”. Journal of Molecular Biology 77 (3): 417–436. doi:10.1016/0022-2836(73)90448-8. PMID 4737866.
- ^ Tulinsky, A.; Van den Hende J.H. (1967). “The crystal and molecular structure of N-brosylmitomycin A”. 米国化学会誌 89 (12): 2905–2911. doi:10.1021/ja00988a018. PMID 6043811.
- ^ Kirby, A. J.; Komarov I.V.; Feeder N. (1998). “Spontaneous, Millisecond Formation of a Twisted Amide from the Amino Acid, and the Crystal Structure of a Tetrahedral Intermediate”. 米国化学会誌 120 (28): 7101–7102. doi:10.1021/ja980700s.
- ^ Evans, D. A.; G. Borg; K. A. Scheidt (2002). “Remarkably Stable Tetrahedral Intermediates: Carbinols from Nucleophilic Additions to N–Acylpyrroles”. アンゲヴァンテ・ケミー 114 (17): 3320–23. doi:10.1002/1521-3757(20020902)114:17<3320::aid-ange3320>3.0.co;2-u.
- ^ a b c d e f g 「ブルース有機化学 第7版 下」p.920
- ^ a b c 「ブルース有機化学 第7版 下」p.924
- ^ Bell, R. P. (1966). “The reversible hydration of carbonyl compounds”. Adv. Phys. Org. Chem. 4 (1): 1–29. doi:10.1016/S0065-3160(08)60351-2.
- ^ Clayden J.; Greeves N.; Warren S.; Wothers P. (2001). Organic Chemistry. Oxford University Press
- ^ 「ブルース有機化学 第7版 下」p.1151
- ^ 「ブルース有機化学 第7版 下」p.921
- ^ Nahm, S.; Weinreb, S. M. (1981). “N-methoxy-N-methylamides as effective acylating agents”. Tetrahedron Letters 22 (39): 3815–18. doi:10.1016/s0040-4039(01)91316-4.
- ^ Adler, M.; Adler S.; Boche G. (2005). “Tetrahedral intermediates in reactions of carboxylic acid derivatives with nucleophiles”. Journal of Physical Organic Chemistry 18 (3): 193–209. doi:10.1002/poc.807.
- ^ 「ブルース有機化学 第7版 下」p.1256
- ^ Babine, R. E.; Bender S. L. (1997). “Molecular Recognition of Protein−Ligand Complexes: Applications to Drug Design”. Chem. Rev. 97 (5): 1359–1472. doi:10.1021/cr960370z. PMID 11851455.
- ^ Bryan, P.; Pantoliano M. W.; Quill S. G.; Hsiao H. Y.; Poulos T. (1986). “Site-directed mutagenesis and the role of the oxyanion hole in subtilisin”. Proc. Natl. Acad. Sci. USA 83 (11): 3743–5. doi:10.1073/pnas.83.11.3743. PMC 323599. PMID 3520553.
参考文献
- Paura Y.Bruice 著、大船泰史・香月勗・西郷和彦・富岡清 訳『ブルース有機化学 第7版[下]』化学同人、2015年2月20日。ISBN 978-4-7598-1585-6。