
重合体

重合体(じゅうごうたい、英: polymer、ポリマー[4][5])は、多数の反復単位からなる高分子(巨大分子)という非常に大きな分子から構成される物質または材料 (en:英語版) である[6]。合成ポリマーも天然ポリマーも、その広範な特性により[7]、日常生活において不可欠かつ遍在的な役割を果たしている[8]。ポリマーは、ポリスチレンのような身近な合成樹脂から、DNAやタンパク質のような生物学的な構造や機能の基礎をなす天然の生体ポリマーまで多岐にわたる。ポリマーは、天然や合成を問わず、モノマーと呼ばれる小分子が多数重合して形成される。その結果、小分子化合物に比べて分子量が大きくなり、強靱性、弾性、粘弾性、(結晶ではなく)非晶質や半結晶構造を形成しやすいなど、特徴のある物理的特性がもたらされる。
「ポリマー(polymer)」という言葉は、ギリシャ語の πολύς(polus, 「多くの、たくさんの」という意味)と μέρος(meros, 「部分」という意味)に由来する。この用語は1833年にイェンス・ヤコブ・ベルセリウスによって作られたが[9]、その定義 (en:英語版) は現代のIUPACの定義とは異なっていた[10]。ポリマーが共有結合した高分子構造であるというという現代的な概念は、1920年にヘルマン・シュタウディンガーによって提唱され[11]、彼はその後10年間をこの仮説の実験的証拠を見つけることに費やした[12]。
ポリマーは、高分子科学(高分子化学と高分子物理学を含む)、生物物理学、材料科学および工学の分野で研究されている。歴史的には、共有化学結合による反復単位の結合から生じる生成物が高分子科学の主な焦点であった。現在では、非共有結合によって形成される超分子ポリマーが、新たに重要な分野として注目されている。ラテックス・ゴムの主成分であるポリイソプレン (en:英語版) は天然ポリマーの例であり、発泡スチロールのポリスチレンは合成ポリマーの例である。生物学的には、本質的にすべての生体高分子、すなわちタンパク質(ポリアミド)、核酸(ポリヌクレオチド)、および多糖は、純粋な高分子であるか、または大部分が高分子成分から構成されている。

一般的な例[編集]

ポリマーには、天然に存在するものと、合成または人工的に作られたものの2種類がある。
天然ポリマー[編集]
麻、シェラック、琥珀、羊毛、絹、天然ゴムなどの天然高分子素材は、何世紀にもわたって使用されてきた。他にも、木材や紙の主成分であるセルロースなど、さまざまな天然ポリマーが存在する。
宇宙ポリマー[編集]
ヘモグリシン(以前はヘモリチンと呼ばれていた)は宇宙ポリマー(space polymer)で、隕石から発見された最初のアミノ酸ポリマーである[13][14][15]。
合成ポリマー[編集]
合成ポリマーの一覧を世界の需要が高い順におおまかに並べると、ポリエチレン、ポリプロピレン、ポリスチレン、ポリ塩化ビニル、合成ゴム、フェノール-ホルムアルデヒド樹脂(またはベークライト)、ネオプレン、ナイロン、ポリアクリロニトリル、PVB、シリコーン、その他多数となる。これらのポリマーは毎年3億3,000万トン以上製造されている(2015年)[16]。
もっとも一般的には、プラスチックの原料となるポリマーの主鎖は、主に炭素原子が連続的に連結して構成している。単純な例としては、ポリエチレンがあり、その反復単位(モノマー)はエチレンである。他にも多くの構造が存在し、たとえばケイ素のような元素はシリコーンのような身近な材料を形成し、シリーパティーや防水性の配管シーリング材などで使用されている。また、酸素は、ポリエチレングリコール、多糖類(グリコシド結合)、DNA(ホスホジエステル結合)などのポリマー骨格にも存在する。
合成[編集]

重合とは、モノマーと呼ばれる小分子を多数結合させ、共有結合でつながった鎖やネットワークを形成するプロセスである。重合のプロセスの際、各々のモノマーから一部の化学基が失われることがある。たとえば、PETポリエステルの重合時にこれが見られる。そのモノマーはテレフタル酸(HOOC - C6H4 - COOH)とエチレングリコール(HO - CH2 - CH2 - OH)であるが、反復単位は - OC - C6H4 - COO - CH2 - CH2 - O - であり、2つの水分子を失った2つのモノマーの組み合わせに相当する。ポリマーに組み込まれる各モノマーの個別の断片は反復単位またはモノマー残基と呼ばれている。
合成法は一般に、段階成長重合(step-growth polymerization、逐次重合とも)と連鎖重合(chain polymerization)の2つに分けられる[17]。両者の本質的な違いは、連鎖重合では、ポリスチレンのようにモノマーが一度に1つずつしか鎖に付加されないのに対し[18]、段階成長重合では、ポリエステルのようにモノマーの連鎖どうしが直接結合できることである[19]。段階成長重合は、それぞれの反応段階ごとに低モル質量の副生成物が生成する縮合重合と重付加に分けられる。

プラズマ重合法のような新しい方法は、どちらのカテゴリーにも属しない。合成重合反応は、触媒の有無にかかわらず行うことができる。生体ポリマー、特にタンパク質の実験室合成は、熱心に研究されている分野である。
生物学的合成[編集]

生体ポリマーには、多糖類、ポリペプチド、ポリヌクレオチドという3つの主要な種類がある。生細胞内でこれらは、DNAポリメラーゼが触媒するDNAの形成など、酵素媒介プロセスにより合成されることがある。タンパク質の合成には、DNAからRNAに遺伝情報を転写し、その情報を翻訳してアミノ酸から特定のタンパク質を合成するという、複数の酵素媒介プロセスが含まれる。このタンパク質は適切な構造と機能を提供するため、翻訳後さらに修飾されることがある。他にも、ゴム、スベリン、メラニン、リグニンなどの生体ポリマーがある。
天然ポリマー改修[編集]
木綿、デンプン、ゴムなどの天然ポリマーは、ポリエチレンやアクリル樹脂などの合成ポリマーが市場に出回るまで、長年に渡って親しまれてきた素材であった。商業的には、重要なポリマーの多くが天然ポリマーの化学的修飾によって合成されている。代表的な例としては、硝酸とセルロースの反応によるニトロセルロースの生成、天然ゴムを硫黄の存在下で加熱することによる加硫ゴムの形成がある。ポリマーを改質する方法には、酸化、架橋、末端キャッピングなどがある。
構造[編集]
高分子材料の構造は、サブナノメートル長(1 nm未満)から巨視的なものまで、さまざまな長さスケールで表すことができる。その構造は実際には階層をなし、それぞれの階層が次の構造の土台となる[20]。ポリマーの構造を表す基点は、構成モノマーの同一性である。次に、微細構造はポリマー内のこれらのモノマーの配列を、基本的な単鎖のスケールで表現する。微細構造はまた、たとえば結晶化、ガラス転移、ミクロ相分離などによって、ポリマーがさまざまな配置で相構造を形成する可能性も決定する[21]。これらの特徴は、ポリマーの物理的および化学的な特性を決定する上で大きな役割を果たす。
モノマーと反復単位[編集]
ポリマーを構成する反復単位(モノマー残基、別名「マー(mer)」)の同一性は、そのポリマーの最初で、最も重要な特性である。ポリマーの命名法は、一般に、ポリマーを構成するモノマー残基の種類に基づいている。1種類の反復単位のみを含むポリマーは同種重合体(ホモポリマー、homopolymer)と呼ばれ、2種類以上の反復単位を含むポリマーは共重合体(コポリマー、copolymer)と呼ばれる[22]。三元(共)重合体(ターポリマー、terpolymer)は、3種類の反復単位を含む共重合体である[23]。
ポリスチレンはスチレン系の反復単位のみから構成され、ホモポリマーに分類される。ポリエチレンテレフタレートは、2つの異なるモノマー(エチレングリコールとテレフタル酸)から合成されるが、反復単位は1種類しか形成しないため、通常はホモポリマーとみなされる。エチレン酢酸ビニルは、2種類以上の反復単位を含んでおり、コポリマーである。生物学的ポリマーの中には、構造的に関連するさまざまな異なるモノマー残基から構成されているものがある。たとえば、DNAのようなポリヌクレオチドは、4種類のヌクレオチドサブユニットから構成されている。
ホモポリマーとコポリマーの例 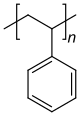
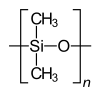
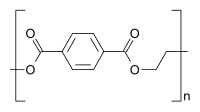
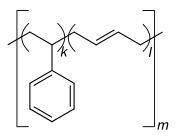
ホモポリマーのポリスチレン ホモポリマーのポリジメチルシロキサン(シリコーン)。主鎖はケイ素原子と酸素原子から構成される。 ホモポリマーのポリエチレンテレフタレートは、反復単位は1種類である。 共重合体のスチレン-ブタジエンゴム:スチレンと1,3-ブタジエンに基づく反復単位が2つ形成され、高分子内では任意の順序で交互に繰り返されるため、ポリマーはランダム共重合体となる。

.svg)


イオン化可能なサブユニット(たとえば、ペンダントカルボキシル基)を含むポリマーは、イオン化可能なユニットの割合が大きい場合は高分子電解質と呼ばれ、小さい場合はアイオノマーと呼ばれる。
微細構造[編集]
ポリマーの微細構造(立体配置、またはコンフィギュレーションと呼ばれることもある)は、鎖の骨格に沿ったモノマー残基の物理的配置に関係している[24]。これらはポリマー構造を構成する要素であり、構造が変化するためには共有結合を切断する必要がある。モノマーや反応条件に応じて、さまざまなポリマー構造が作り出される。分岐していない鎖を1本だけ含む直鎖状高分子から構成されるポリマーもある。非分岐ポリエチレンの場合、この鎖は長鎖 n-アルカンである。主鎖と側鎖とを持つ分岐高分子もあり、ポリエチレンの場合、側鎖はアルキル基である。特に非分岐高分子では、固体状態では半結晶となる場合があり、下の図では結晶鎖の部分が赤色で強調表示されている。
分岐ポリマーおよび非分岐ポリマーは通常熱可塑性プラスチックであるが、多くのエラストマーは「主鎖」の間に広い網目状の架橋を持つ。一方、密な網目状の架橋は熱硬化性につながる。図では、架橋と分岐が赤点で示されている。高度に分岐したポリマーは非晶質であり、固体中の分子はランダムに相互作用する。




ポリマーの構造[編集]

ポリマーの微細構造における重要な特徴は、その構造と形状であり、これは分岐点が単純な直鎖からの逸脱をもたらす方法に関係している[25]。分岐ポリマー分子は、1つ以上の置換基を持つ側鎖または分岐を持つ主鎖で構成される。分岐ポリマーの種類には、星型ポリマー、櫛型ポリマー、ポリマーブラシ、デンドロナイズドポリマー(デンドロン化ポリマー)、はしご型ポリマー、デンドリマーなどがある[25]。トポロジー的に平面的な反復単位から構成される二次元ポリマー(2DP)も存在する。ポリマーの構造は、溶液粘度、溶融粘度、さまざまな溶媒への溶解性、ガラス転移温度、溶液中の個々の高分子コイルのサイズなど、その物理的特性の多くに影響を及ぼす。さまざまな構造を持つ高分子材料を合成するために、たとえばリビング重合 (en:英語版) など、さまざまな技術を採ることができる。
鎖長[編集]
鎖の長さを表す一般的な手段は重合度であり、これは鎖に組み込まれたモノマーの数を定量化したものである[26][27]。他の分子と同様に、ポリマーの大きさを分子量で表すこともできる。合成重合技術では通常、鎖長の統計的分布が得られ、分子量は加重平均で表される。数平均分子量(Mn)と重量平均分子量(Mw)が最も一般的に報告されている[28][29]。この2つの値の比(Mw / Mn)が分散度(Đ)であり、一般に分子量分布の幅を表すために使用される[30]。
ポリマーの物理的性質は[31]、ポリマー鎖の長さ(または相当する分子量)に強く依存する[32]。分子量の物理的影響の重要な例として、ポリマー溶融物(融液)の粘度(流動抵抗)のスケーリングがある[33]。重量平均分子量()が溶融粘度()に及ぼす影響は、そのポリマーが絡み合い分子量を上回るか下回るかによって異なる。絡み合い分子量以下では となり[要説明]、絡み合い分子量以上では となる。後者の場合、ポリマーの鎖長を10倍にすると粘度は1,000倍以上に増加する[34][要ページ番号]。さらに鎖長を長くすると、鎖の運動性が低下し、強度と靭性が増し、ガラス転移温度(Tg)が上昇する傾向がある[35]。これは、鎖長が長くなるのにつれて、ファンデルワールス引力や絡み合いなどの鎖間相互作用が増加する結果である[36][37]。これらの相互作用は、個々の鎖の位置をより強固に固定して、より高い応力とより高い温度の両面で、変形やマトリクスの破壊に抵抗する傾向がある。




共重合体のモノマー配列[編集]
共重合体は、統計共重合体(ランダム共重合体)、交互共重合体、ブロック共重合体、グラフト共重合体、グラジエント共重合体(傾斜共重合体)のいずれかに分類される。次の模式図では Ⓐ と Ⓑ が2つの反復単位を表している。

ランダム共重合体
グラジエント共重合体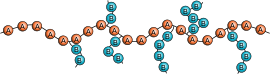
グラフト共重合体
交互共重合体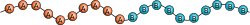
ブロック共重合体

- 交互共重合体(alternating copolymers)は、2つのモノマー残基が規則的に交互に配列している([AB]nの誤植ではない)[38]。たとえば、フリーラジカル連鎖成長重合によって形成されるスチレンと無水マレイン酸の等モル共重合体があげられる[39]。ナイロン66のような段階成長共重合体も、厳密にはジアミン残基と二酸残基の交互共重合体と考えることができるが、アミンと酸の二量体残基を反復単位とするホモポリマーと表現されることが多い[40]。
- 周期共重合体(periodic copolymers)は、3種類以上のモノマー単位が規則正しく配列している[41]。
- 統計共重合体(statistical copolymers)は、モノマー残基が統計的規則に従って配列している。鎖の特定の位置に特定の種類のモノマー残基が存在する確率が、周囲のモノマー残基の種類に依存しないランダム共重合体は、真のランダム共重合体(truly random copolymer)と呼ばれることがある[42][43]。 たとえば、塩化ビニルと酢酸ビニルの連鎖成長共重合体はランダムである[39]。
- ブロック共重合体(block copolymers)は、異なるモノマー単位が長く配列している[39][40]。2種類の化学種(たとえばAとB)の2つまたは3つのブロックを持つポリマーは、それぞれジブロック共重合体およびトリブロック共重合体と呼ばれる。それぞれ異なる化学種(たとえばA、B、C)の3つのブロックを持つポリマーはトリブロックターポリマーと呼ばれる。
- グラフト共重合体(graft copolymers)は、主鎖とは異なる組成や配置の反復単位を持つ側鎖や分岐を含む。分岐は、あらかじめ形成された主鎖の高分子に付加される[39]。
共重合体中のモノマーを、さまざまな方法で主鎖に沿って組織化することができる。モノマー配列が制御された共重合体を、配列制御ポリマーと呼ぶ[44]。交互共重合体、周期共重合体、およびブロック共重合体は、配列制御ポリマーの簡単な例である。
立体規則性[編集]
立体規則性(tacticity)は、高分子内で隣接する構造単位におけるキラル中心の相対的な立体化学を表す。立体規則性には3種類があり、イソタクチック(すべての置換基が同じ側にある)、シンジオタクチック(置換基が交互に配置される)、およびアタクチック(置換基がランダムに配置される)である。

イソタクチック
シンジオタクチック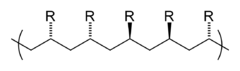
アタクチック(すなわちランダム)



形態学[編集]
一般に、高分子形態学(Polymer morphology)では、空間におけるポリマー鎖の配列とマイクロスケールでの秩序を研究する。ポリマーの巨視的な物理的特性は、ポリマー鎖間の相互作用と関連している。
 ランダムに配向したポリマー |
 複数のポリマーの連結 |
- 無秩序ポリマー: アタクチックポリマー、高分岐ポリマー、ランダム共重合体は、固体状態ではアモルファス(非晶質、すなわちガラス状構造)を形成する[45]。溶融状態や溶液状態では、ポリマーは絶えず変化して「統計クラスター」を形成する傾向がある(自由連結鎖モデルを参照)。固体状態では、分子のそれぞれの立体構造は凍結している。鎖状分子の引っ掛かりや絡み合いにより、鎖の間に「機械的結合」が生じる。分子間および分子内の引力は、分子セグメントが互いに十分に接近している部位でのみ生じる。分子が不規則な構造をとるため、狭い範囲での配置が阻害される。
 ポリエチレン: 分子が密に詰まったジグザグ構造 |
 結束分子を持つラメラ |
 球晶 |
 ポリプロピレンのらせん構造 |
 p-アラミド:赤い点線は水素結合 |
- 線状ポリマー: 周期構造を持ち、分岐が少なく、立体規則性がある(たとえばアタクチックでない)線状ポリマーは、固体状態では半結晶構造を持つ[45]。単純なポリマー(ポリエチレンなど)では、鎖は結晶内にジグザグ構造で存在する。いくつかのジグザグ構造では、微結晶(結晶子、ラメラとも)と呼ばれる高密度な鎖の塊を形成する。ラメラはポリマーの長さよりもはるかに小さく、約10 nmであることが多い[46]。これらは1本または複数の分子鎖がおおむね規則的に折りたたまれることで形成される。ラメラとラメラの間には非晶質構造が存在する。個々の分子はラメラ間の絡み合いをもたらし、2つ(またはそれ以上)のラメラ(結束分子(tie molecule)と呼ばれる鎖)の形成に関与することもある。複数のラメラが球晶と呼ばれる上位構造を形成し、その直径は0.05 - 1 mmの範囲が多い[46]。
反復単位の(機能)残基の種類や配置は、結晶化度や副原子価結合の強さに影響し、そして決定する。イソタクチックポリプロピレンでは、分子はらせんを形成している。ジグザグ構造と同様に、このようならせんは高密度な鎖の詰め込みを可能にする。p-アラミドの場合のように反復単位の残基が水素結合の形成する場合、特に強い分子間相互作用が生じる。強い分子内会合が形成されると、回路トポロジーが異なる一本鎖の多様な折りたたみ状態を形成することがある。結晶化度と上位構造は常にその形成条件に依存している(ポリマーの結晶化を参照)。非晶質構造に比べ、半結晶構造はポリマーの剛性、密度、溶融温度、および抵抗力を高める。
- 架橋ポリマー: 網目の広い架橋ポリマーはエラストマーであり、(熱可塑性樹脂とは異なり)溶融することはない。架橋ポリマーを加熱しても分解するだけである。一方、熱可塑性エラストマー (en:英語版) は可逆的な「物理的架橋」をしており、加熱すると溶融する。ブロック共重合体は熱可塑性エラストマーの一種であり、ハードセグメントが結晶化しやすく、ソフトセグメントが非晶質構造を持ち、ハードセグメントが広い網目状の物理的架橋を行う。
 網目の広い架橋ポリマー(エラストマー) |
 エラストマーに応力がかけられた場合 |
 「架橋部位」としての微結晶 (en:英語版) : 熱可塑性エラストマーの一種 |
 引張応力下の半結晶性熱可塑性エラストマー |
結晶化度[編集]
結晶性(crystalline)という用語は、ポリマーに適用される場合、やや曖昧である。場合によっては、結晶性という用語は従来の結晶学と同じ使われ方をする。たとえば、X線結晶構造解析用に調製されたサンプルのような結晶性タンパク質やポリヌクレオチドの構造は、セル寸法が数百オングストローム以上の1つまたは複数のポリマー分子から構成される従来の単位セルの観点で定義されることがある。合成ポリマーは、(高分子ではなく)原子長スケールの三次元秩序を持つ領域を含む場合、大まかに結晶性と表現することができ、これらの領域は通常、隣接する鎖の分子内折りたたみや積み重なりから生じる。合成ポリマーは、結晶性領域と非晶質領域の両方から構成されることがある。その場合、結晶化度(degree of crystallinity)は、結晶性物質の重量分率または体積分率で表すことができる。完全に結晶性の合成ポリマーはほとんどない[47]。ポリマーの結晶性は結晶化度によって特徴付けられ、その範囲は、完全に非結晶性のポリマーを示す0から、理論的に完全に結晶性のポリマーを示す1まである。微結晶領域を持つポリマーは一般に、完全な非晶質ポリマーよりも強靭で(壊れることなく、より多く曲げることができる)、衝撃にも強くなる[48]。結晶化度が0または1に近いポリマーは透明になる傾向があり、結晶化度が中間のポリマーは結晶領域またはガラス領域による光散乱のために不透明になる傾向がある。多くのポリマーでは、結晶化度は透明度の低下とも関連している。
分子鎖立体構造[編集]
ポリマー分子が占める空間は、一般に、鎖の質量中心から鎖自体までの平均距離である回転半径で表される。あるいは、ポリマー鎖が占める浸透体積の観点から表すこともでき、これは回転半径の3乗に比例する[49]。溶融した非晶質状態のポリマーの最も単純な理論モデルは、理想鎖である。
特性[編集]
ポリマーの特性はその構造に依存し、物理的基盤によって分類される。ポリマーが連続的な巨視的物質としてどのように振る舞うかは、多くの物理的特性や化学的特性で説明される。これらはバルク物性あるいは熱力学に従った示強性に分類される。
機械的特性[編集]

ポリマーのバルク特性は、最終用途で最も注目される特性である。これらは、巨視的スケールでポリマーが実際にどのような挙動を示すかを決定づける特性である。
引張強度[編集]
材料の引張強度は、材料が破断するまでにどれだけの伸びに耐えられるかを定量化したものである[50][51]。これは、ポリマーの物理的強度や耐久性に依存する用途では非常に重要である。たとえば、引張強度がより高いゴムバンドは、破断する前により大きな重量に耐えることができる。一般に、引張強度はポリマー鎖の長さと架橋度によって増加する。
ヤング率[編集]
ヤング率はポリマーの弾性を定量化したものである。これは、ひずみが小さい場合において、ひずみに対する応力の変化率の比として定義される。引張強度と同様に、これはポリマーの物理的性質が重視される用途(ゴムバンドなど)では非常に重要である。弾性率は温度に大きく依存する。粘弾性は、複雑な時間依存の弾性応答を説明し、荷重が取り除かれると応力-ひずみ曲線にヒステリシスを示す。動的機械分析(DMA)は、荷重を振動させ、その結果生じるひずみを時間の関数として測定することにより、この複素弾性率を測定する。
輸送特性[編集]
拡散性などの輸送特性は、分子が高分子マトリックス中を移動する速さを表す。こうした特性は、フィルムや膜などのポリマーの多くの用途において非常に重要である。
個々の高分子の移動は、レプテーションと呼ばれるプロセスによって起こり、それぞれの鎖状分子は隣接する鎖との絡み合いにより拘束をうけ、仮想チューブ内を移動する。レプテーション理論によって、ポリマー分子のダイナミクスや粘弾性を説明することができる[52]。
相挙動[編集]
結晶化と融解[編集]
化学構造によって、ポリマーは半結晶か非晶質のいずれかの状態になる。半結晶性ポリマーは結晶化と融解転移を起こす可能性があるが、非結晶性ポリマーはそうではない。ポリマーにおける結晶化や融解は、水や他の分子流体の場合のような固液相転移を示唆するものではない。その代わりに、結晶化と融解は2つの固体状態(すなわち、半結晶と非晶質)の間の相転移を意味する。結晶化は、ガラス転移温度(Tg)以上、融解温度(Tm)以下で起こる。
ガラス転移[編集]
すべてのポリマー(非晶質または半結晶)はガラス転移を起こす。ガラス転移温度(Tg)は、ポリマーの製造、加工、使用にとってきわめて重要な物理的パラメータである。Tg 以下では分子運動が停止し、ポリマーは脆く、ガラス状になる。Tg を超えると分子運動が活性になり、ポリマーはゴムのような粘性を持つ。ガラス転移温度は、ポリマーの分岐や架橋の程度を変えたり、可塑剤を添加することで操作することができる[53]。
結晶化と融解が一次相転移であるのに対し、ガラス転移はそうではない[54]。ガラス転移は、二次相転移の特徴(図に示すような熱容量の不連続性など)を共有しているが、一般的に平衡状態間の熱力学的転移とはみなされない。
混合挙動[編集]

一般に、ポリマー混合物は、小分子材料の混合物よりもはるかに混和性が低くなる。この効果は、通常、混合の原動力が相互作用エネルギーよりもむしろエントロピーであるという事実から生じる。言い換えれば、混和性の物質が溶液を形成するのは通常、互いの相互作用が自己相互作用よりも有利であるためではなく、各成分が利用できる体積の増加に伴うエントロピーの増加、すなわち自由エネルギーの増加のためである。このエントロピーの増加は、混合される粒子数(またはモル数)に比例する。ポリマー分子は小分子よりもはるかに大きく、したがって一般に比体積(比容)が大きいため、ポリマー混合物に含まれる分子の数は、同じ体積の小分子混合物に含まれる分子の数よりもはるかに少なくなる。一方、混合のエネルギーは、高分子混合物と小分子混合物では体積あたりで同等である。このため、ポリマー溶液の混合自由エネルギーは増大し、それにより溶媒和が不利になる傾向がある。その結果、ポリマーの濃縮溶液は小分子溶液よりも遙かに希少にある。
さらに、ポリマー溶液や混合物の相挙動は、小分子混合物よりも複雑である。ほとんどの小分子溶液が、冷却時に相分離する上部臨界溶液温度相転移(UCST)のみを示すのに対し、ポリマー混合物は一般に、加熱時に相分離する下部臨界溶液温度相転移(LCST)を示す。
希薄溶液では、ポリマーの特性は溶媒とポリマーの相互作用によって特徴づけられる。良溶媒中では、ポリマーは膨潤して、大きな体積を占めるように見える。このシナリオでは、溶媒とモノマー反復単位間の分子間力が、分子内相互作用よりも支配的である。貧溶媒では、分子内力が支配的となり、鎖は収縮する。シータ溶媒(θ溶媒)、すなわち第2ビリアル係数の値が0になるポリマー溶液の状態では、ポリマー - 溶媒間の分子間斥力とモノマー - モノマー間の分子内引力がちょうど釣り合う。θ条件(フローリー条件とも呼ばれる)では、ポリマーは理想的なランダムコイルのように振る舞う。これらの状態間の転移は、コイル・グロビュール転移として知られている。
可塑剤の含有[編集]
可塑剤の添加は、ガラス転移温度 Tg を低下させ、ポリマーの柔軟性を増加させる傾向がある。また、ガラス転移温度 Tg の冷却速度への依存性も変化する[55]。可塑剤の分子が水素結合を形成すると、鎖の移動性はさらに変化する。可塑剤は一般的に、ポリマーと化学的に類似した小分子で、ポリマー鎖の間に隙間を作ることで移動性を高め、鎖間相互作用を低減させる。可塑剤がどのように作用するかを表す好例としてポリ塩化ビニル(PVC)があげられる。無可塑ポリ塩化ビニル(uPVC)はパイプなどの原料に使われる。パイプは強度と耐熱性を維持する必要があるため、可塑剤を含まない。可塑化ポリ塩化ビニルは、柔軟性を持たせるために衣料品に使われる。また可塑剤は、ポリマーをより柔軟にするために、ある種の粘着フィルムにも含まれている。
化学的性質[編集]
ポリマー鎖間の引力は、ポリマーの特性を決定する上で大きな役割を果たす。ポリマー鎖は非常に長いため、1分子毎にこのような鎖間の相互作用が多く存在しており、通常の分子間の引力に比べて、ポリマー特性への影響が増幅される。ポリマーのさまざまな側鎖基が、ポリマー自身の鎖間にイオン結合や水素結合を持つことがある。これらの強い力は、一般に高い引張強度と高い結晶融点をもたらす。
ポリマー内の分子間力は、モノマー単位内の双極子によって影響を受けることがある。アミド基やカルボニル基を持つポリマーは、隣接する鎖間で水素結合を形成することができる。ある鎖のN-H基で部分的に正に帯電した水素原子は、別の鎖のC=O基の部分的に負に帯電した酸素原子に強く引き寄せられる。このような強い水素結合は、たとえば、ウレタンや尿素結合を含むポリマーの高い引張強度と融点をもたらす。ポリエステルは、C=O基の酸素原子とH-C基の水素原子との間に双極子-双極子結合を持つ。双極子結合は水素結合ほど強くないため、ポリエステルの融点と強度はケブラー(トワロン)よりも低いが、ポリエステルは柔軟性に優れている。ポリエチレンのように非極性のモノマー単位を持つポリマーは、弱いファンデルワールス力によってのみ相互作用する。その結果、溶融温度は他のポリマーよりも一般に低くなる。
市販の塗料や接着剤のように、ポリマーが液体に分散または溶解している場合、化学的性質や分子間相互作用が溶液の流れ方に影響を与え、自己集合化によってポリマーが複雑な構造になることもある。ポリマーをコーティングとして塗布する場合、化学的性質はコーティングの接着性や、耐水性をもつ超疎水性ポリマーコーティングのような外部材料との相互作用に影響する。概して、ポリマーの化学的特性は、新しい高分子材料製品の設計において重要な要素である。
光学的性質[編集]
PMMAやHEMA:MMA共重合体などのポリマーは、固体色素レーザー(固体色素ドープポリマーレーザーとも呼ばれる)の利得媒質のマトリックスとして使用される。これらのポリマーは高い表面品質をもち、透明度も高いため、レーザー特性は高分子マトリックス中に分散するレーザー色素によって支配される。このタイプのレーザーは有機レーザーの種類に属し、非常に狭い線幅が得られることで知られており、分光法や分析用途に有用である[56]。レーザー用途に使用されるポリマーの重要な光学パラメータは、温度による屈折率の変化であり、dn/dTとしても知られている。ここに取り上げたポリマーの場合、297 ≤ T ≤ 337 Kの範囲において、(dn/dT) ~ −1.4×10−4 [K−1]である[57]。
電気的特性[編集]
ポリエチレンのような従来のポリマーのほとんどは電気絶縁体であるが、π共役結合を含むポリマーの開発により、ポリチオフェンなどのポリマー系半導体が豊富になった。このため、有機エレクトロニクス (en:英語版) の分野で多く応用されている。
用途[編集]
今日、合成ポリマーは生活のほとんど全ての分野で使用されている。これらがなければ、現代社会はまったく違ったものになっただろう。ポリマーが広く利用されているのは、低密度、低コスト、優れた断熱性/電気絶縁性、高い耐腐食性、製造エネルギーの低さ、最終製品への加工の容易さといった、独特な特性に関係している。特定の用途に応じて、複合材料のように他の材料と組み合わせることで、ポリマーの特性を調整したり強化することができる。ポリマーの使用により、エネルギーの節約(自動車や航空機の軽量化、建物の断熱化)、食品や飲料水の保護(包装)、土地の節約と肥料の使用削減(合成繊維)、他の材料の保護(コーティング)、人命の保護と救助(衛生、医療用途)を可能にする。代表的な用途の一部を次にあげる。
- 衣料品、スポーツウェア、アクセサリー:
ポリエステルやポリ塩化ビニルの衣料品、スパンデックス、スポーツシューズ、ウェットスーツ、サッカーボールやビリヤードボール、スキーやスノーボード、ラケット、パラシュート、帆、テントやシェルター。 - 電子および光技術:
有機電界効果トランジスタ(OFET)、有機発光ダイオード(OLED)、太陽電池、テレビ部品、コンパクトディスク(CD)、フォトレジスト、ホログラフィー。 - 包装および容器:
フィルム、ボトル、食品包装、樽 - 絶縁材:
電気絶縁および断熱材、スプレーフォーム - 建築および構造用途:
ガーデン家具、ポリ塩化ビニル窓、床材、シーリング、パイプ - 塗料、接着剤、潤滑剤:
ワニス、接着剤、分散剤、落書き防止コーティング、防汚コーティング (en:英語版) 、非粘着性表面コーティング、潤滑剤 - 自動車部品:
タイヤ、バンパー、フロントウィンドウ、ワイパー、燃料タンク、カーシート - 家庭用品:
バケツ、キッチンウェア、玩具(工作セットやルービックキューブなど)。 - 医療用途:
血液バッグ、注射筒、ゴム手袋、縫合糸、コンタクトレンズ、人工関節、薬物送達および放出制御、細胞増殖用マトリックス。 - 対人衛生とヘルスケア:
高吸水性ポリマーを使用したおむつ、歯ブラシ、化粧品、シャンプー、コンドーム。 - 防護:
個人用防護具、防弾チョッキ、宇宙服、ロープ - 分離技術:
合成膜、燃料電池膜、ろ過膜、イオン交換樹脂 - 貨幣:
ポリマー紙幣、決済カード - 3Dプリンティング
標準命名法[編集]
ポリマー物質に命名するためのいくつかの規則がある。消費者向け製品に見られるような、一般的なポリマーの多くは、一般名または慣用名で呼ばれている。慣用名は、標準化された命名規則ではなく、歴史的な先例や一般的な用法に基づいて付与される。米国化学会(ACS)[58]と国際純正・応用化学連合(IUPAC)[59]はともに標準化された命名規則を提案して。これらの規則は類似しているが、同一ではない[60]。いくつかの命名規則の相違する例を次の表に示す。
| 一般名 | ACS名 | IUPAC名 |
|---|---|---|
| Poly(ethylene oxide) or PEO | Poly(oxyethylene) | Poly(oxyethylene) |
| Poly(ethylene terephthalate) or PET | Poly(oxy-1,2-ethanediyloxycarbonyl-1,4-phenylenecarbonyl) | Poly(oxyethyleneoxyterephthaloyl) |
| Nylon 6 or Polyamide 6 | Poly[imino(1-oxo-1,6-hexanediyl)] | Poly[azanediyl(1-oxohexane-1,6-diyl)] |
どちらの標準化規則でも、ポリマーの名前は、反復単位の正確な性質よりも、合成元のモノマーを反映することを意図している(出所に基づく命名法)。たとえば、単純なアルケンであるエテン(ethene)から合成されるポリマーはポリエテン(polyethene)と呼ばれ、重合プロセスで二重結合が取り除かれても接尾辞 -ene は残る。
 →
→


しかしながら、IUPACの構造命名法では、優先的構成反復単位の命名に基づいている[61]。
特性評価[編集]
ポリマーの特性評価には、化学組成、分子量分布、物理的特性を決定するための多くの技術が使われている。一般的な手法を次にあげる。
- サイズ排除クロマトグラフィー(ゲル浸透クロマトグラフィー (en:英語版) とも呼ばれる)は、静的光散乱を併用する場合もあり、数平均分子量、重量平均分子量、分散度を測定することができる。
- 静的光散乱や小角中性子散乱などの散乱技術は、溶液中または融液中の高分子の大きさ(回転半径)を測定するために使用される。これらの技術は、ミクロ相分離したブロック共重合体、ポリマーミセル、その他の材料の三次元構造の特性を把握するためにも使用される。
- 広角X線散乱(広角X線回折とも呼ばれる)は、ポリマーの結晶構造(またはその欠如)を決定するために使用される。
- フーリエ変換赤外分光法、ラマン分光法、核磁気共鳴分光法などの分光法技術を使用して、化学組成を決定することができる。
- 示差走査熱量測定法は、ガラス転移温度、結晶化温度、融解温度などのポリマーの熱特性の特性の把握に使用される。ガラス転移温度は、動的機械分析によっても求めることができる。
- 熱重量分析は、ポリマーの熱安定性を評価するのに有用な手法である。
- レオロジーは、流動と変形挙動の特徴を把握するために使用される。粘度、弾性率、その他のレオロジー特性の測定に使用できる。レオロジーはまた、分子構造(分子量、分子量分布、分岐)を決定し、ポリマーの加工方法を理解するためにもよく使用される。
劣化[編集]

ポリマーの劣化とは、熱、光、特定の化学物質、酸素、酵素など、1つまたは複数の環境要因の影響下で、ポリマーまたはポリマー系製品の特性(引張強度、色、形状、分子量)が変化することである。このような物性の変化は、多くの場合、ポリマー骨格の結合破壊の結果であり、分子鎖の末端や鎖内の任意の位置で起こりうる。
このような変化は望ましくないことが多いが、ときには生分解やリサイクルのように、環境汚染を防ぐことを目的としている場合もある。また、分解は生物医学的な場面でも有用である。たとえば、ポリ乳酸とポリグリコール酸の共重合体は、創傷を縫合した後にゆっくりと分解する加水分解性縫合糸に使用されている。
ポリマーの分解しやすさはその構造に依存する。エポキシや芳香族官能基を含む鎖は、特に紫外線による分解を受けやすく、また、ポリエステルは加水分解による劣化を受けやすい。不飽和骨格を含むポリマーは、オゾンクラッキングによって劣化する。炭素系ポリマーは、ポリジメチルシロキサンのような無機高分子よりも熱劣化が起こりやすく、そのためほとんどの高温用途には適さない。
ポリエチレンの劣化は、ポリマーの原子を結合している結合が無作為に切れるランダム切断によって起こる。ポリエチレンは、450 °C以上に加熱すると分解して炭化水素の混合物を形成する。また、分子鎖末端の切断の場合、モノマーが放出され、このプロセスはアンジッピング(unzipping)または解重合(depolymerization)と呼ばれる。どの機構が支配的かは、ポリマーの種類や温度に依存する。一般に、反復単位に小さな置換基を持たないか、1つしか持たないポリマーは、ランダム鎖切断によって分解する。
リサイクル目的でのポリマー廃棄物の分別では、プラスチックの種類を識別するために米国プラスチック産業会が開発した樹脂識別コードを使用することで容易にすることができる。
ポリマー製品の故障[編集]

安全上重要なポリマー部品の故障は、ポリマー製の燃料配管の亀裂や劣化による火災など、重大事故につながる可能性がある。特に1990年代の米国で、アセタール樹脂製の配管継手やポリブチレン管の塩素による亀裂により、住宅で多くの深刻な浸水を引き起こした。水道水中の微量の塩素が配管のポリマーを劣化させる問題は、部品の押出成形や射出成形が不十分な場合に急速に起こった。成形不良のアセタール継手が侵され、応力が集中する継手のねじ山に沿って亀裂が入った。

ポリマーの酸化は、医療機器でも事故を引き起こしている。最も古くから知られている故障モードの一つがオゾンクラッキング(亀裂)で、これは天然ゴムやニトリルゴムなどの影響を受けやすいエラストマーで、オゾンが攻撃する際の分子鎖切断によって起こる。これらのゴムは、反復単位に二重結合を含んでおり、オゾン酸化によって切断される。燃料配管に亀裂が入るとチューブ断面を貫通し、燃料漏れの原因となりうる。エンジンルーム内でクラックが発生すると、電気火花がガソリンに引火し、重大な火災を引き起こす可能性がある。医療用途では、ポリマーの劣化が埋め込み型器具の物理的および化学的特性の変化をもたらす可能性がある[62]。
ナイロン6,6は酸による加水分解を受けやすく、ある事故では燃料配管の破断によって軽油が道路に流出した。軽油が道路に流出すると、堆積物がブラックアイスのように滑りやすくなるため、後続車の事故を引き起こす可能性がある。さらに、軽油がアスファルト合材からアスファルテンを溶出させるため、アスファルトコンクリート路面が損傷し、アスファルト路面の劣化と道路の構造的完全性が損なわれる。
歴史[編集]
人類が誕生して以来、ポリマーは、日常生活に欠かせないものであった。羊毛(ケラチン)や、木綿・麻の繊維(セルロース)を衣服に、カミガヤツリ(セルロース)を紙に使用したことは、古代社会がどのようにポリマーを原材料に工芸品を作ったかを示す一例にすぎない。パラゴムノキのラテックス樹液(天然ゴム)は、オルメカ、マヤ、アステカがボウルや防水布、容器の材料として使用し始め、ずっと後の16世紀になって、南米を経てヨーロッパに到達した[63]。
ポリマーの化学的な操作は19世紀までさかのぼるが、当時はまだその性質は理解されていなかった。ポリマーの挙動は、当初、トーマス・グレアムが提唱した理論によって説明された。グレアムは、ポリマーを未知の力によって結合した小分子のコロイド状凝集体と考えていた。
理論的な知識が不足していたにもかかわらず、革新的で入手しやすく安価な材料を供給するポリマーの可能性はすぐに理解された。アンリ・ブラコノー、アレクサンダー・パークス、フリードリヒ・ヴィルヘルム・リューダースドルフ、ナサニエル・ヘイワードをはじめとする多くの研究者たちによる天然ポリマーの改質に関する研究は、この分野における多くの重要な進歩を決定づけた[64]。彼らの貢献により、セルロイド、ガラリス、パークシン、レーヨン、加硫ゴム、そして後にはベークライトなどの材料が発見された。これらの材料は、すぐに工業的な製造工程に組み入れられ、衣料品(生地、ボタンなど)、食器、装飾品として家庭に普及した。
1920年、ヘルマン・シュタウディンガーが『Über Polymerisation(重合)』という重要な論文を発表し[65]、ポリマーは共有結合で連結した原子の長い鎖であると提唱した。彼の研究は長い間議論されたが、最終的には科学界に受け入れられた。この業績により、シュタウディンガーは1953年にノーベル賞を受賞した[66]。
1930年代以降、ポリマーは全盛を迎え、新しい種類のポリマーが発見され、急速に天然素材に代わって商業的用途が見いだされた。その開発は、強力な経済力を持つ産業分野によって推進され、より安価な原料からの革新的なモノマーの合成、より効率的な重合プロセス、ポリマーの特性評価技術の向上、およびポリマーの高度な理論的理解などに貢献した幅広い学術コミュニティによって支えられてきた[64]。

1953年以降、生体高分子の研究を除いて、高分子科学の分野で6つのノーベル賞が授与されている。このことは、高分子科学が現代の科学技術に大きな影響を与えたことを証明している。1980年、トッド卿は「重合の発展は、おそらく化学が成し遂げた最大の出来事であり、日常生活に最も大きな影響を与えたものであろう」と総括している[68]。
参考項目[編集]
脚注[編集]
- ^ Roiter, Y.; Minko, S. (2005). “AFM Single Molecule Experiments at the Solid-Liquid Interface: In Situ Conformation of Adsorbed Flexible Polyelectrolyte Chains”. Journal of the American Chemical Society 127 (45): 15688–15689. doi:10.1021/ja0558239. PMID 16277495.
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). オンライン版: (2006-) "polymer".
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). オンライン版: (2006-) "macromolecule (polymer molecule)".
- ^ “Polymer – Definition of polymer”. The Free Dictionary. 2013年7月23日閲覧。
- ^ "Define polymer". Dictionary Reference. 2013年7月23日閲覧。
- ^ “Polymer on Britannica”. 2023年8月11日閲覧。
- ^ Painter, Paul C.; Coleman, Michael M. (1997). Fundamentals of polymer science: an introductory text. Lancaster, Pa.: Technomic Pub. Co.. p. 1. ISBN 978-1-56676-559-6
- ^ McCrum, N. G.; Buckley, C. P.; Bucknall, C. B. (1997). Principles of polymer engineering. Oxford; New York: Oxford University Press. p. 1. ISBN 978-0-19-856526-0
- ^ If two substances had molecular formulae such that one was an integer multiple of the other – e.g., acetylene (C2H2) and benzene (C6H6) – Berzelius called the multiple formula "polymeric". See: Jöns Jakob Berzelius (1833) "Isomerie, Unterscheidung von damit analogen Verhältnissen" (Isomeric, distinction from relations analogous to it), Jahres-Bericht über die Fortschitte der physischen Wissenschaften …, 12: 63–67. From page 64: "Um diese Art von Gleichheit in der Zusammensetzung, bei Ungleichheit in den Eigenschaften, bezeichnen zu können, möchte ich für diese Körper die Benennung polymerische (von πολυς mehrere) vorschlagen." (In order to be able to denote this type of similarity in composition [which is accompanied] by differences in properties, I would like to propose the designation "polymeric" (from πολυς, several) for these substances.)
Originally published in 1832 in Swedish as: Jöns Jacob Berzelius (1832) "Isomeri, dess distinktion från dermed analoga förhållanden," Årsberättelse om Framstegen i Fysik och Kemi, pages 65–70; the word "polymeriska" appears on page 66. - ^ Jensen, William B. (2008). “Ask the Historian: The origin of the polymer concept”. Journal of Chemical Education 85 (5): 624–625. Bibcode: 2008JChEd..85..624J. doi:10.1021/ed085p624. オリジナルの2018-06-18時点におけるアーカイブ。 2013年3月4日閲覧。.
- ^ Staudinger, H (1920). “Über Polymerisation [On polymerization]” (ドイツ語). Berichte der Deutschen Chemischen Gesellschaft 53 (6): 1073–1085. doi:10.1002/cber.19200530627.
- ^ Allcock, Harry R.; Lampe, Frederick W.; Mark, James E. (2003). Contemporary Polymer Chemistry (3 ed.). Pearson Education. p. 21. ISBN 978-0-13-065056-6
- ^ McGeoch, J.E.M.; McGeoch, M.W. (2015). “Polymer amide in the Allende and Murchison meteorites.”. Meteoritics & Planetary Science 50 (12): 1971–1983. Bibcode: 2015M&PS...50.1971M. doi:10.1111/maps.12558.
- ^ McGeogh, Julie E. M.; McGeogh, Malcolm W. (28 September 2022). “Chiral 480nm absorption in the hemoglycin space polymer: a possible link to replication”. Scientific Reports 12 (1): 16198. doi:10.1038/s41598-022-21043-4. PMC 9519966. PMID 36171277.
- ^ Staff (2021年6月29日). “Polymers in meteorites provide clues to early solar system”. Science Digest 2023年1月9日閲覧。
- ^ “World Plastics Production”. 2023年8月11日閲覧。
- ^ Sperling, L. H. (Leslie Howard) (2006). Introduction to physical polymer science. Hoboken, N.J.: Wiley. p. 10. ISBN 978-0-471-70606-9
- ^ Sperling, p. 11
- ^ Sperling, p. 15
- ^ Sperling, p. 29
- ^ Bower, David I. (2002). An introduction to polymer physics. Cambridge University Press. ISBN 9780511801280
- ^ Rudin, p.17
- ^ Cowie, p.4
- ^ Sperling, p. 30
- ^ a b Rubinstein, Michael; Colby, Ralph H. (2003). Polymer physics. Oxford; New York: Oxford University Press. p. 6. ISBN 978-0-19-852059-7
- ^ McCrum, p. 30
- ^ Rubinstein, p. 3
- ^ McCrum, p. 33
- ^ Rubinstein, pp. 23–24
- ^ Painter, p. 22
- ^ De Gennes, Pierre Gilles (1979). Scaling concepts in polymer physics. Ithaca, N.Y.: Cornell University Press. ISBN 978-0-8014-1203-5
- ^ Rubinstein, p. 5
- ^ McCrum, p. 37
- ^ Introduction to Polymer Science and Chemistry: A Problem-Solving Approach By Manas Chanda
- ^ O'Driscoll, K.; Amin Sanayei, R. (July 1991). “Chain-length dependence of the glass transition temperature”. Macromolecules 24 (15): 4479–4480. Bibcode: 1991MaMol..24.4479O. doi:10.1021/ma00015a038.
- ^ Pokrovskii, V. N. (2010). The Mesoscopic Theory of Polymer Dynamics. Springer Series in Chemical Physics. 95. Bibcode: 2010mtpd.book.....P. doi:10.1007/978-90-481-2231-8. ISBN 978-90-481-2230-1
- ^ Edwards, S. F. (1967). “The statistical mechanics of polymerized material”. Proceedings of the Physical Society 92 (1): 9–16. Bibcode: 1967PPS....92....9E. doi:10.1088/0370-1328/92/1/303.[リンク切れ]
- ^ Painter, p. 14
- ^ a b c d Rudin p.18-20
- ^ a b Cowie p.104
- ^ Periodic copolymer. International Union of Pure and Applied Chemistry. (2014). doi:10.1351/goldbook.P04494 2020年4月9日閲覧。.
- ^ Painter, p. 15
- ^ Sperling, p. 47
- ^ Lutz, Jean-François; Ouchi, Makoto; Liu, David R.; Sawamoto, Mitsuo (2013-08-09). “Sequence-Controlled Polymers” (英語). Science 341 (6146): 1238149. doi:10.1126/science.1238149. ISSN 0036-8075. PMID 23929982.
- ^ a b Bernd Tieke: Makromolekulare Chemie. 3. Auflage, Wiley-VCH, Weinheim 2014, S. 295f (in German).
- ^ a b Wolfgang Kaiser: Kunststoffchemie für Ingenieure. 3. Auflage, Carl Hanser, München 2011, S. 84.
- ^ Sayed, Abu (August 2014). Types of polymer: Requirements of fibre forming polymer.
- ^ Allcock, Harry R.; Lampe, Frederick W.; Mark, James E. (2003). Contemporary Polymer Chemistry (3 ed.). Pearson Education. p. 546. ISBN 978-0-13-065056-6
- ^ Rubinstein, p. 13
- ^ Ashby, Michael; Jones, David (1996). Engineering Materials (2 ed.). Butterworth-Heinermann. pp. 191–195. ISBN 978-0-7506-2766-5
- ^ Meyers, M. A.; Chawla, K. K. (1999). Mechanical Behavior of Materials. Cambridge University Press. p. 41. ISBN 978-0-521-86675-0. オリジナルの2013-11-02時点におけるアーカイブ。 2018年12月31日閲覧。
- ^ Fried, Joel R. (2003). Polymer Science & Technology (2nd ed.). Prentice Hall. pp. 155–6. ISBN 0-13-018168-4
- ^ Brandrup, J.; Immergut, E.H.; Grulke, E.A. (1999). Polymer Handbook (4 ed.). Wiley-Interscience. ISBN 978-0-471-47936-9
- ^ Meille, S.; Allegra, G.; Geil, P. et al. (2011). “Definitions of terms relating to crystalline polymers (IUPAC Recommendations 2011)”. Pure and Applied Chemistry 83 (10): 1831–1871. doi:10.1351/PAC-REC-10-11-13 2018年12月31日閲覧。.
- ^ Capponi, S.; Alvarez, F.; Racko, D. (2020), “Free Volume in a PVME Polymer–Water Solution”, Macromolecules XXX (XXX): XXX-XXX, Bibcode: 2020MaMol..53.4770C, doi:10.1021/acs.macromol.0c00472, hdl:10261/218380
- ^ Duarte, F. J. (1999). “Multiple-prism grating solid-state dye laser oscillator: optimized architecture”. Applied Optics 38 (30): 6347–6349. Bibcode: 1999ApOpt..38.6347D. doi:10.1364/AO.38.006347. PMID 18324163.
- ^ Duarte, F. J. (2003). Tunable Laser Optics. New York: Elsevier Academic. ISBN 978-0122226960
- ^ CAS: Index Guide, Appendix IV ((c) 1998)
- ^ IUPAC (1976). “Nomenclature of Regular Single-Strand Organic Polymers”. Pure Appl. Chem. 48 (3): 373–385. doi:10.1351/pac197648030373.
- ^ Wilks, E.S.. “Macromolecular Nomenclature Note No. 18”. 2003年9月25日時点のオリジナルよりアーカイブ。2023年8月11日閲覧。
- ^ Hiorns, R. C.; Boucher, R. J.; Duhlev, R.; Hellwich, Karl-Heinz; Hodge, Philip; Jenkins, Aubrey D.; Jones, Richard G.; Kahovec, Jaroslav et al. (2012-10-03). “A brief guide to polymer nomenclature (IUPAC Technical Report)” (英語). Pure and Applied Chemistry 84 (10): 2167–2169. doi:10.1351/PAC-REP-12-03-05. ISSN 0033-4545.
- ^ Iakovlev, V.; Guelcher, S.; Bendavid, R. (August 28, 2015). “Degradation of polypropylene in vivo: A microscopic analysis of meshes explanted from patients”. Journal of Biomedical Materials Research Part B: Applied Biomaterials 105 (2): 237–248. doi:10.1002/jbm.b.33502. PMID 26315946.
- ^ Hurley, Paul E. (May 1981). “History of Natural Rubber” (英語). Journal of Macromolecular Science: Part A - Chemistry 15 (7): 1279–1287. doi:10.1080/00222338108056785. ISSN 0022-233X.
- ^ a b Feldman, Dorel (January 2008). “Polymer History” (英語). Designed Monomers and Polymers 11 (1): 1–15. doi:10.1163/156855508X292383. ISSN 1568-5551.
- ^ Staudinger, H. (1920-06-12). “Über Polymerisation”. Berichte der Deutschen Chemischen Gesellschaft (A and B Series) 53 (6): 1073–1085. doi:10.1002/cber.19200530627. ISSN 0365-9488.
- ^ “The Nobel Prize in Chemistry 1953” (英語). NobelPrize.org. 2020年6月25日閲覧。
- ^ Feldman, Dorel (2008-01-01). “Polymer History”. Designed Monomers and Polymers 11 (1): 1–15. doi:10.1163/156855508X292383.
- ^ “Lord Todd: the state of chemistry”. Chemical & Engineering News Archive 58 (40): 28–33. (1980-10-06). doi:10.1021/cen-v058n040.p028. ISSN 0009-2347.
参考書[編集]
- Cowie, J. M. G. (John McKenzie Grant) (1991). Polymers: chemistry and physics of modern materials. Glasgow: Blackie. ISBN 978-0-412-03121-2
- Hall, Christopher (1989). Polymer materials (2nd ed.). London; New York: Macmillan. ISBN 978-0-333-46379-6
- Rudin, Alfred (1982). The elements of polymer science and engineering. Academic Press. ISBN 978-0-12-601680-2
- Wright, David C. (2001). Environmental Stress Cracking of Plastics. RAPRA. ISBN 978-1-85957-064-7
外部リンク[編集]
この記事の外部リンクはウィキペディアの方針やガイドラインに違反しているおそれがあります。 |