
ファイル:Phylogenetic tree of coronaviruses.jpg
高解像度版はありません。
Phylogenetic_tree_of_coronaviruses.jpg (571 × 451 ピクセル、ファイルサイズ: 75キロバイト、MIME タイプ: image/jpeg)
ウィキメディア・コモンズのファイルページにある説明を、以下に表示します。
|
{kind=link}
{kind=link}
{kind=link}
{kind=link}
概要
| 解説 |
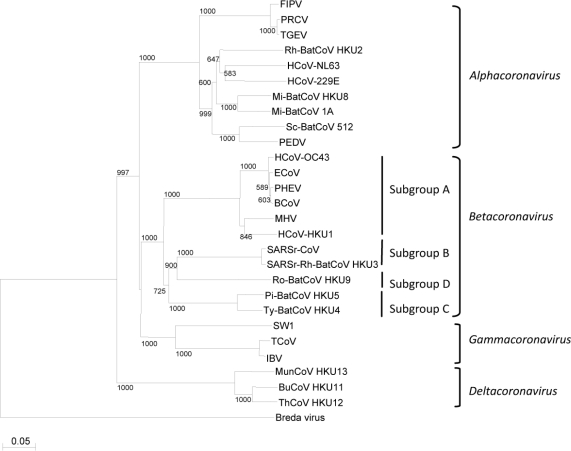
English: Phylogenetic analysis of RNA-dependent RNA polymerases (Pol) of coronaviruses with complete genome sequences available. The tree was constructed by the neighbor-joining method and rooted using Breda virus polyprotein. Bootstrap values were calculated from 1000 trees. 1118 amino acid positions in Pol were included. The scale bar indicates the estimated number of substitutions per 20 amino acids. All abbreviations for the coronaviruses were the same as those in Figure 1. |
| 日付 | |
| 原典 | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ |
| 作者 | Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen |
ライセンス
このファイルはクリエイティブ・コモンズ 表示 3.0 非移植ライセンスのもとに利用を許諾されています。
- あなたは以下の条件に従う場合に限り、自由に
- 共有 – 本作品を複製、頒布、展示、実演できます。
- 再構成 – 二次的著作物を作成できます。
- あなたの従うべき条件は以下の通りです。
- 表示 – あなたは適切なクレジットを表示し、ライセンスへのリンクを提供し、変更があったらその旨を示さなければなりません。これらは合理的であればどのような方法で行っても構いませんが、許諾者があなたやあなたの利用行為を支持していると示唆するような方法は除きます。
ファイルの履歴
過去の版のファイルを表示するには、その版の日時をクリックしてください。
| 日付と時刻 | サムネイル | 寸法 | 利用者 | コメント | |
|---|---|---|---|---|---|
| 現在の版 | 2020年3月7日 (土) 02:42 | | 571 × 451 (75キロバイト) | Guest2625 | Uploaded a work by Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ with UploadWizard |
ファイルの使用状況
このファイルを使用しているページはありません。
グローバルなファイル使用状況
以下に挙げる他のウィキがこの画像を使っています:
- bn.wikipedia.org での使用状況
- en.wikipedia.org での使用状況
- fa.wikipedia.org での使用状況
- fr.wikipedia.org での使用状況
- ig.wikipedia.org での使用状況
- sq.wikipedia.org での使用状況
- su.wikipedia.org での使用状況
{kind=link}