
ホウ素化
金属触媒を用いたC-H結合のホウ素化反応(英: Metal-catalyzed C–H borylation reaction)とは、遷移金属によって触媒される有機化学反応であり、脂肪族化合物もしくは芳香族化合物のC–H結合を活性化して有機ホウ素化合物に変換する反応である[1]。金属触媒を用いたC-Hホウ素化反応は遷移金属を用いてC–H結合をC–B結合に直接変換する反応である。この変換は一般的なホウ素化に比べ、安くて豊富に存在する炭化水素を出発物質として、有機化合物の官能化(prefunctionalization)を抑え、毒性のある副生成物の生成を抑制しつつ、生理活性を有した化学物質の合成を進めることができる[2][3]。ボロン酸とボロン酸エステルが有機分子のホウ素化によく用いられるホウ素源である[4]。ボロン酸は3価のホウ素を含む有機化合物で、1つのアルキル置換基と2つのヒドロキシ基を持っている。同様に、ボロン酸エステルは1つのアルキル基と2つのエステル基を持っている。ボロン酸とボロン酸エステルはホウ素原子に結合しているアルキル基の種類(アルキル、アルケニル、アルキニル、アリールなど)によって分類される。最もよく用いられる出発物質はボロン酸エステルを含む化合物で、一般式(RO)2B-B(OR)2で表される。例えば、ビス(ピナコラート)ジボロン(B2Pin2)や、ビス(カテコラート)ジボロン(B2Cat2)などである[5]。

ボロン酸もしくはボロン酸エステルのホウ素原子はsp2混成であり、空のp軌道が存在する。このため、ボロン酸やボロン酸エステルはルイス酸として機能する。ボロン酸やボロン酸エステルのC–B結合はC–C単結合よりやや長く、1.55-1.59 Åである。C–B結合の結合長がC–C結合より長いので、C-Bの結合エネルギーはC–C結合のそれよりやや小さい(C-B結合:323 kJ/mol、C-C結合:358 kJ/mol)[6]。 The 炭素-水素結合の結合長は約1.09 Åであり、結合エネルギーは約413 kJ/molである。ゆえにC–B結合は反応性の低いC-H結合を置き換える中間体として有用である。
有機ホウ素化合物は炭素-ホウ素結合を含む有機化合物である。有機ホウ素化合物は、C-B結合がC–X (X = Br,Cl)、C–O、C–N、C–C結合に容易に変換できるため、有機合成化学において幅広い用途がある。このため、C-B結合を分子に取り込む多くの方法が開発されている[7]。有機ホウ素化合物は古典的にはグリニャール試薬からヒドロホウ素化もしくは二ホウ素化を経由して合成される[8]。ホウ素化反応はこの代替法を示している。
金属触媒を用いたC–Hホウ素化反応[編集]
脂肪族C–Hホウ素化[編集]
この反応はハートウィッグによって報告された。アルカンはCp*Rh(η4-C6Me6)を触媒として用いることで高選択的に1級のC–H結合をホウ素化することができる[9]。特筆すべきは、この触媒は炭化水素鎖にヘテロ原子が存在する場合でも1級C-H結合を選択的にホウ素化することである。ロジウム触媒を用いたメチル基のC-Hのホウ素化はヘテロ原子の位置に関わらず化学選択性が発現する。ホウ素化の選択性は立体的なかさ高さや電子密度にほとんどよらずに発現し、アセタールやエーテル、アミン、フッ化アルキルなど様々な化合物で現れる[10]。さらに、シクロヘキサンなどのように1級のC-H結合が存在しない基質を用いた場合反応は起こらない。

一級アルカンの選択的官能化が優先的に起こるのは、速度論的にも熱力学的にも1級のアルキル-金属錯体が2級のそれより好ましいからである[11]。

2級アルキル錯体より1級アルキル錯体が安定である理由はいくつかある。まず、1級アルキル錯体は立体的にすいているため基質と金属が接近しやすい。次に、部分負電荷は多くの場合金属-アルキル錯体のα-炭素に存在するため、2級アルキル配位子より1級アルキル配位子の方が電荷を負担する能力が高いためである。 ロジウム錯体を使用した際の脂肪族のC-Hホウ素化の選択性の由来は水素-重水素交換質量分析によって明らかになった。H/D で置き換えて反応させると位置選択性およびメタル-アルキル中間体の官能化位置選択性が低下した[12]。

合成において、C-Hホウ素化はポリマーをホウ素化ののち酸化してヒドロキシ化するのに利用されている[13]。

芳香族化合物のC–Hホウ素化[編集]
位置選択的なC-Hホウ素化[編集]
不活性なC-H結合(ベンゼン)の触媒的ホウ素化の最初の例はIr(Cp*)(H)(Bpin)を触媒として用いる反応で、スミスとイバーソン(Iverson)によって報告された。しかしこの変換の触媒効率は悪く、120 h、150 °Cで3回しか反応させることができなかった[14]。ハートウィッグや彼の共同研究者による研究の結果、効率的かつ実用的なアレーンのホウ素化が実現した。芳香族C–Hホウ素化はハートウィッグと石山竜生らは4,4’-ジ-tert-ブチルジピリジン (dtbpy)と[Ir(COD)(OMe)]2を触媒としてジボロン試薬であるビス(ピナコラート)ジボロンをホウ素源として用い、[15]。この触媒系匂いては芳香族のC-H結合のホウ素化は出発となるアレーンの立体障害のために、立体選択的に進行する。この芳香族C-H結合官能化における選択性は一般的にC-H結合でo-置換体が存在しない場合、C-B結合でもo-置換体が生成する反応は起こらないという規則が存在する[11]。官能基が1つだけ存在する場合、メタ位とパラ位で置換が起こり、メタ置換体とパラ置換体の比が2:1となる。オルト異性体は立体効果のため生成しない[16]。

1,2-置換または1,4-置換の芳香族化合物の場合、ホウ素の付加は1箇所でしか起こらない。1,3-置換の芳香族化合物では、立体的に付加できる水素原子が1つしかないため、やはり1箇所でのみ選択的に反応する。

これは、電気的な効果によって選択性が決まる芳香族求電子置換反応とは対照的である[17]。

芳香族C-Hホウ素化の合成における重要性は以下の図に示した。1,3-置換芳香族化合物は1,3,5-有機ホウ素化合物に変換でき、ホウ素を様々な官能基へと変換することができる[15]。

芳香族C–H官能化はコンプラナジンA(10-ヒドロキシリコポジウムアルカロイド)の全合成に利用されている。コンプラナジンAはヒトのグリア細胞で神経成長因子(NGF)をつくり、mRNAで発現させる。コンプラナジンAは新しい神経ネットワークをつくるため、アルツハイマー病の薬として注目されている天然化合物である[18]。コンプラナジンAはハートウィッグと石山竜生によって開発された芳香族C–H結合のホウ素化と鈴木・宮浦カップリングを組み合わせ、その後でBoc保護基を外して合成された。

複素芳香族化合物のC-Hホウ素化[編集]
複素環式芳香族化合物(ヘテロアレーン)もイリジウム触媒下でホウ素化することができる。しかしこの場合ベンゼン環と違って位置選択性に電子効果が影響する。このような化合物の例としてフラン、ピロール、チオフェンなどがある。このような化合物ではヘテロのα位にあるC-H結合で反応が起こる。この位置で反応が起こるのは、ここのプロトンの酸性がもっとも高いからである[11]。

オルト位のC-Hホウ素化[編集]
立体配置を決定する置換基のない化合物で触媒によりホウ素化の位置を選択する方法が開発されている。例えば、ボーベル(Boebel)とハートウィッグはジメチルヒドロシリルを含む芳香族化合物は、イリジウム触媒下でC-H結合がシラン基に対してo-ホウ素化することを見出している[19]。ヒドロシリルを用いるとオルト位に選択的に反応するのは、Si-H結合が金属中心に可逆的に付加するためで、これがヒドロシリル化合物のオルト位におけるC-H結合の優先的な開裂に繋がる。他にも多くの官能基を用いた芳香族化合物のオルト位選択的なC-H結合のホウ素化法が開発されている[20][21][22]。

芳香族C-Hホウ素化の反応機構[編集]
芳香族化合物のC-Hホウ素化の反応中間体はトリスボリル-イリジウムであると考えられている速度論解析と同位体標識研究によって触媒サイクルにおいてイリジウム(III)-トリスボリル錯体が芳香族化合物と反応することがわかっている[23]。 触媒サイクルのバリエーションとして下記に示すようなヒドロシラン化合物のオルト位ホウ素化がある。速度論解析のデータから観察された、シクロオクテンに配位したトリスボリル錯体は速やかに解離し、16電子錯体をつくる。芳香族化合物としてベンジルジメチルシランが用いられた場合、ベンジルジメチルシランがトリスボリルイリジウム触媒と反応して、Si-H結合が金属中心に可逆的に付加すると考えられている。その後、酸化的付加および還元的脱離により、オルト位のC–H結合が選択的に活性化されると考えられている[24]。

メタ位選択的ホウ素化: メタ位選択的なC-Hホウ素化は合成上重要な反応であり、2002年にミシガン州立大学のスミスIII世によって発見された。しかしこの反応は1,3-二置換ベンゼンでしか進行しなかった。約12年後、インドにあるCBMR(Centre of Biomedical Research)のチャトパディヤイのチームはメタ位選択的なC-H活性化・ホウ素化の新たな手法を発見した。彼のチームは同じ基質を用い、配位子を変えることによって位置選択性が変わることを示した。メタ選択性の発現は2つのパラメーター1) 静電気的相互作用, 2) 2級B-N相互作用 によって変化することがわかった[25]。
同時期に、東大薬学院の金井求は2級相互作用によるメタ選択的ホウ素化のコンセプトを提唱している[26]。
有機ホウ素化合物による還元反応[編集]
コーリー・バクシ・柴田還元[編集]
1981年、平尾明、伊津野真一、中浜精一、山崎升らはプロキラルな芳香族ケトンをキラルなアミノアルコールとボランを用いて不斉還元すると、対応する2級アルコールを60% eeで得られることを報告した。彼らはキラルなアミノアルコールがボランと反応してアルコキシ-アミン-ボラン錯体を形成することを発見した。この錯体は比較的剛直な五員環であり、熱や加水分解に安定で多くのプロトン性溶媒、非プロトン性溶媒に可溶である[27]。
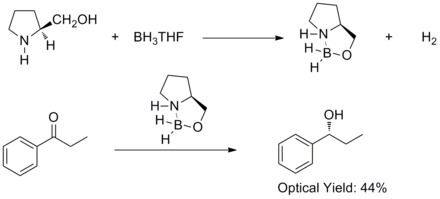
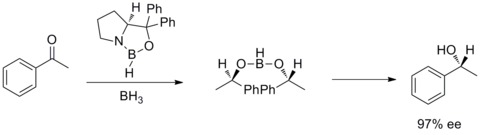
1987年には、イライアス・コーリー、バクシ、柴田らはボランとキラルなアミノアルコールからオキサザボロリジンが生成することを発見した。そしてオキサザボロリジンはプロキラルなケトンをBH3THFの存在下、高エナンチオ選択的に還元することがわかった。このケトンの不斉還元はコーリー・バクシ・柴田還元(CBS還元)と呼ばれている[28][29]
ミッドランド還元[編集]
1977年、M. M. ミッドランドらは(+)-α-ピネンを9-BBNでヒドロホウ素化して得られるB-3-α-ピナニル-9-ボラビシクロ [3,3,1] ノナン(B-3-α-ピナニル-9-BBN)がベンザルアルデヒド-α-dを(S)-(+)-ベンジル-α-dアルコールに量論的に不斉還元する[30]。
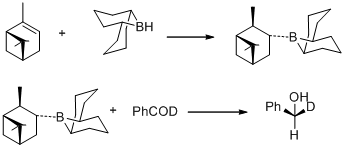
同年、M. M.ミッドランドは(+)-α-ピネンと9-BBNを反応させて容易に得られるB-3-α-ピナニル-9-BBNを還元剤として用いることができることを発見した。新しい還元剤はのちにシグマ・アルドリッチより発売され、アルピンボランと命名された。アルピンボランを使ったカルボニル基の不斉還元はミッドランド還元(ミッドランドアルピンボラン還元)と呼ばれる[31]。
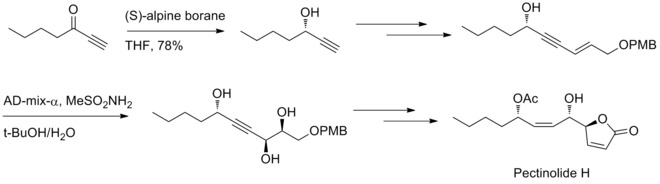
2012年、U. R. Y. ベンケイツワール(Venkateswarlu)らはペクチノリドH(pectinolide H)の立体選択的な合成法を開発した。ミッドランド還元とシャープレス不斉ジヒドロキシ化がC–4’, C–5とC–1’の3つの不斉中心の合成に利用されている[32]。
有機ホウ素化合物を使用したカップリング反応[編集]
ペタシス反応[編集]
1993年、N. A.ペタシスとI. Akrltopoulouはマンニッヒ反応を修正することで効率的なアリルアミンの合成法を開発した。修正されたマンニッヒ反応では、ビニルボロン酸が求核剤として反応し不斉なアリルアミンを与える[33][34]。
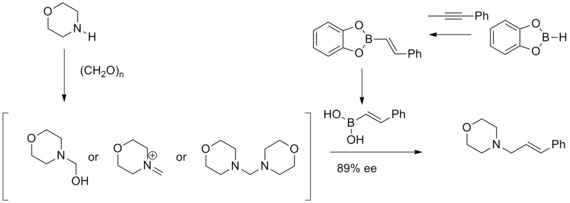
ラウシュの不斉アリル化[編集]
1978年、R. W. ホフマンとT.ヘロルトはキラルなアリルボロンエステルから2級ホモアリルアルコールをエナンチオ選択的に合成する方法を報告した。ホモアリルアルコールは高収率かつ高エナンチオ選択的に得られた[35]。
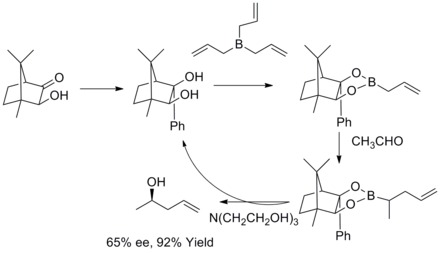
1985年、W. R.ラウシュらはアリルボロン酸酒石酸エステルをキラルまたはアキラルなアルデヒドと面選択的に反応させる方法を発見した。後年、W.R.ラウシュらはこの方法をcis-2-ブテン-1,4-ジオールやアンチジオールの合成に適用した。この種の反応はラウシュの不斉アリル化として知られている[36][37][38][39]。
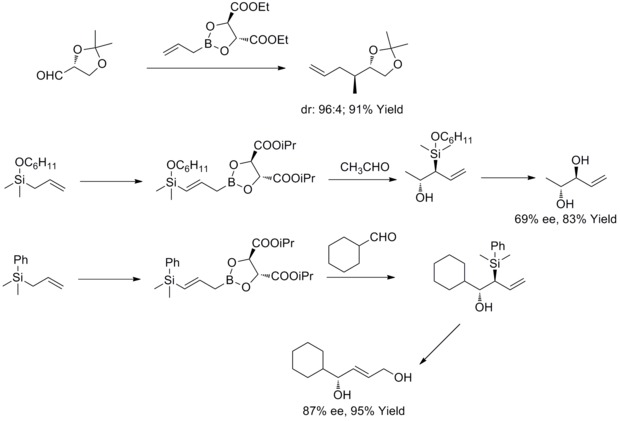
2011年、R. A. フェルナンデスとP. カッタングル(Kattanguru)は(8S, 11R, 12R)- および (8R, 11R, 12R)-トプセントリド(topsentolide) B2のジアステレオマーを8段階で全合成することに成功した。この論文中で、ジアステレオ選択的なラウシュのアリル化反応が2つのキラル中間体を導入する際に用いられた。それから著者らは2つのジアステレオマーを2つのキラル中間体を経由することで合成した[40]。

鈴木・宮浦カップリング[編集]
1979年、宮浦憲夫と鈴木章が塩化アリールとアルキル-1-エニルボランをテトラキス(トリフェニルホスフィン)パラジウム(0)と塩基を触媒にして反応させ、アリール化(E)アルケンを高収率で得たと報告した。その後鈴木章らはこの反応を他の有機ホウ素化合物と他のアルケニル、アルキル、アリール、ハロゲン化アリール、トリフラートにも適用した。この有機ホウ素化合物と有機ハロゲン化物を反応させるパラジウム触媒カップリング反応は鈴木・宮浦カップリングとして知られている[41][42]。
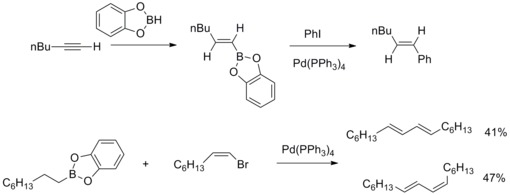
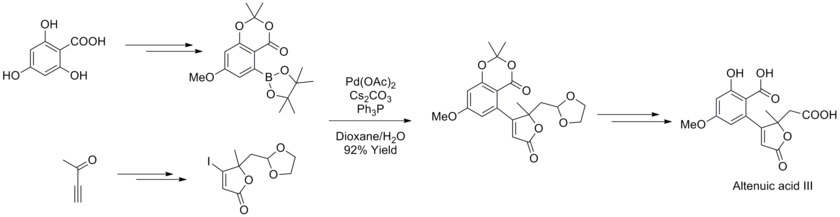
2013年、ヨアキム・ポドレック(Joachim Podlech)らはアルテルナリア マイコトキシン アルテン酸 IIIの構造をNMRで決定し、その全合成を達成した。合成においては多くの官能基をもつボロン酸化合物とブテノライドを鈴木・宮浦カップリングで反応させ、天然化合物の前駆体を高収率で得た[43]。
改良ウルマン反応によるビアリールエーテルおよびビアリールアミンの合成[編集]
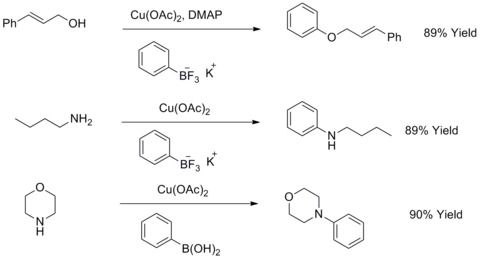
1904年、フリッツ・ウルマンはハロゲン化アリールとフェノールを縮合させてビアリールエーテルを作る反応が銅粉によって促進されることを発見した。この反応はウルマン反応ないしはウルマン縮合と呼ばれている。1906年、I.ゴールドバーグ(I. Goldberg)はこの反応を、ハロゲン化アリールとアミドを塩化銅(I)および炭酸カリウムの存在下で反応させてアリールアミンを合成する反応へと拡張した。この反応はゴールドバーグ修正版ウルマン縮合と呼ばれている[44]。2003年、R. A.ベイティとT. D.コークはこの反応に一部修正を加え、脂肪族アルコール、アミンまたはアニリンと三フッ化ボロン酸カリウムを反応させ、アリールエーテルやアリールアミンを合成する手法を開発した[45][46]。
関連項目[編集]
脚注[編集]
- ^ Hartwig, John F. (2012). “Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations”. Accounts of Chemical Research 45 (6): 864–873. doi:10.1021/ar200206a. ISSN 0001-4842. PMID 22075137.
- ^ Cho, J. Y.; Tse, M. K.; Holmes, D.; Maleczka, R. E., Jr.; Smith, M. R. Science (Washington D.C.) 2002, 295, 305.
- ^ Ishiyama, T.; Nobuta, Y.; Hartwig, J. F.; Miyaura, N. Chem. Commun. 2003, 2924.
- ^ Brown, H. C.; Kramer, G. W.; Levy, A. B.; Midland, M. M. Organic Synthesis via Boranes;Wiley-Interscience: New York, 1975; Vol. 1.
- ^ Braunschweig, H.; Guethlein, F. Angew. Chem. Int. Ed. 2011, 50, 12613-12616.
- ^ Hall, D. G. (2011) Structure, Properties, and Preparation of Boronic Acid Derivatives, in Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials (Volume 1 and 2), Second Edition (ed D. G. Hall), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany. doi: 10.1002/9783527639328.ch1
- ^ C−H Activation for the Construction of C−B Bonds Ibraheem A. I. Mkhalid, Jonathan H. Barnard, Todd B. Marder, Jaclyn M. Murphy, and John F. Hartwig Chemical Reviews 2010 110 (2), 890-931 doi:10.1021/cr900206p
- ^ Wade, L. G., Organic Chemistry. Upper Saddle River: Pearson Education, Inc., 2010.
- ^ Chen, H.; Schlecht, S.; Semple, T. C.; Hartwig, J. F. Science 2000, 287, 1995-1997.
- ^ Lawrence, J. D.; Takahashi, M.; Bae, C.; Hartwig, J. F. J. Am. Chem. Soc. 2004, 126, 15334-15335.
- ^ a b c Regioselectivity of the borylation of alkanes and arenes Hartwig, J.F. Chemical Society Reviews 2011, 40, 1992. doi:10.1039/C0CS00156B
- ^ Wei, C. S.; Jimenez-Hoyos, C. A.; Videa, M.F.; Hartwig, J. F.; Hall, M. B. J. Am. Chem. Soc., 2010, 132, 3078.
- ^ Kondo, Y.; Garcia-Cuadrado, D.; Hartwig, J.F.; Boaen, N.K.; Wagner, N.L.; Hillmyer, M.A. J. Am. Chem. Soc. 2002, 124, 1164. doi:10.1021/ja016763j
- ^ Iverson, Carl N.; Smith, Milton R. (1999-08-06). “Stoichiometric and Catalytic B−C Bond Formation from Unactivated Hydrocarbons and Boranes” (英語). Journal of the American Chemical Society 121 (33): 7696–7697. doi:10.1021/ja991258w.
- ^ a b Hartwig, J.F. Accounts of Chemical Research 2012, 45, 864-873.
- ^ Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N.; Hartwig, J.F. J. Am. Chem. Soc., 2002, 124, 390-391.
- ^ Liskey, C. Iridium-Catalyzed Borylation of Aromatic and Aliphatic C–H bonds: Methodology and Mechanism. Dissertation, University of Illinois. Urbanan-Champaign. 2013.
- ^ Fischer, D.F; Sarpong, R. J. Am. Chem. Soc. 2010, 132, 5926-5927.
- ^ Boebel, T. A.; Hartwig, J. F. J. Am. Chem. Soc. 2008, 130, 7534.
- ^ Ishiyama, T.; Miyaura, N.; Isou, H.; Kikuchi, T. Chem. Commun. 2010, 46, 159.
- ^ Kawamorita, S.; Ohmiya, H.; Hara, K.; Fukuoka, A.; Sawamura, M. J. Am. Chem. Soc., 2009, 131, 5058.
- ^ Ros, A.; Estepa, B.; Lopez-Rodriquez, R.; Alvarez, E.; Fernandez, R.; Lassaletta, J.M. Angew. Chem. Int. Ed. 2011, '50', 1.
- ^ Boller, T.M.; Murphy, J. M.; Hapke, M.; Ishiyama, T.; Miyaura, N.; Hartwig, J.F. J. Am. Chem. Soc. 2005, 127, 14263.
- ^ Boebel, T.A.; Hartwig, J.F. J. Am. Chem. Soc., 2008, 130, 7534.
- ^ Bisht, R.; Chattopadhyay, B. J. Am. Chem. Soc. 2016, 138, 84.
- ^ Kanai et al. Nat. Chem. 2015, 7, 712.
- ^ Hirao, Akira; Itsuno, Shinichi; Nakahama, Seiichi; Yamazaki, Noboru (1981). “Asymmetric reduction of aromatic ketones with chiral alkoxy-amineborane complexes”. Journal of the Chemical Society, Chemical Communications (7): 315. doi:10.1039/c39810000315.
- ^ Corey, E. J.; Bakshi, Raman K.; Shibata, Saizo (September 1987). “Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications”. Journal of the American Chemical Society 109 (18): 5551–5553. doi:10.1021/ja00252a056.
- ^ Corey, E. J.; Bakshi, Raman K.; Shibata, Saizo; Chen, Chung Pin; Singh, Vinod K. (December 1987). “A stable and easily prepared catalyst for the enantioselective reduction of ketones. Applications to multistep syntheses”. Journal of the American Chemical Society 109 (25): 7925–7926. doi:10.1021/ja00259a075.
- ^ Midland, M.Mark; Tramontano, Alfonso; Zderic, Stephen A (July 1977). “The facile reaction of B-alkyl-9-borabicyclo[3.3.1]nonanes with benzaldehyde”. Journal of Organometallic Chemistry 134 (1): C17–C19. doi:10.1016/S0022-328X(00)93625-8.
- ^ Midland, M. Mark; Tramontano, Alfonso; Zderic, Stephen A. (June 1977). “Preparation of optically active benzyl-.alpha.-d alcohol via reduction by B-3.alpha.-pinanyl-9-borabicyclo[3.3.1]nonane. A new highly effective chiral reducing agent”. Journal of the American Chemical Society 99 (15): 5211–5213. doi:10.1021/ja00457a068.
- ^ Ramesh, D.; Shekhar, V.; Chantibabu, D.; Rajaram, S.; Ramulu, U.; Venkateswarlu, Y. (March 2012). “First stereoselective total synthesis of pectinolide H”. Tetrahedron Letters 53 (10): 1258–1260. doi:10.1016/j.tetlet.2011.12.122.
- ^ Petasis, Nicos A.; Akritopoulou, Irini (January 1993). “The boronic acid mannich reaction: A new method for the synthesis of geometrically pure allylamines”. Tetrahedron Letters 34 (4): 583–586. doi:10.1016/S0040-4039(00)61625-8.
- ^ Yu, Tao; Li, Hui; Wu, Xinyan; Yang, Jun (2012). “Progress in Petasis Reaction”. Chinese Journal of Organic Chemistry 32 (10): 1836. doi:10.6023/cjoc1202092.
- ^ Herold, Thomas; Hoffmann, Reinhard W. (October 1978). “Enantioselective Synthesis of Homoallyl Alcoholsvia Chiral Allylboronic Esters”. Angewandte Chemie International Edition in English 17 (10): 768–769. doi:10.1002/anie.197807682.
- ^ Roush, William R.; Walts, Alan E.; Hoong, Lee K. (December 1985). “Diastereo- and enantioselective aldehyde addition reactions of 2-allyl-1,3,2-dioxaborolane-4,5-dicarboxylic esters, a useful class of tartrate ester modified allylboronates”. Journal of the American Chemical Society 107 (26): 8186–8190. doi:10.1021/ja00312a062.
- ^ Roush, William R.; Ando, Kaori; Powers, Daniel B.; Halterman, Ronald L.; Palkowitz, Alan D. (January 1988). “Enantioselective synthesis using diisopropyl tartrate modified (E)- and (Z)-crotylboronates: Reactions with achiral aldehydes”. Tetrahedron Letters 29 (44): 5579–5582. doi:10.1016/S0040-4039(00)80816-3.
- ^ Roush, William R.; Grover, Paul T. (January 1990). “Diisopropyl tartrate (E)-γ-(dimethylphenylsilyl)allylboronate, a chiral allylic alcohol β-carbanion equivalent for the enantioselective synthesis of 2-butene-1,4-diols from aldehydes”. Tetrahedron Letters 31 (52): 7567–7570. doi:10.1016/S0040-4039(00)97300-3.
- ^ Roush, William R.; Gover, Paul T.; Lin, Xiaofa (January 1990). “Diisopropyl tartrate modified (E)-γ-[(cyclohexyloxy)dimethylsilyl-allylboronate, a chiral reagent for the stereoselective synthesis of anti 1,2-diols via the formal α-hydroxyallylation of aldehydes”. Tetrahedron Letters 31 (52): 7563–7566. doi:10.1016/S0040-4039(00)97299-X.
- ^ Fernandes, Rodney A.; Kattanguru, Pullaiah (November 2011). “Total synthesis of (8S,11R,12R)- and (8R,11R,12R)-topsentolide B2 diastereomers and assignment of the absolute configuration”. Tetrahedron: Asymmetry 22 (20–22): 1930–1935. doi:10.1016/j.tetasy.2011.10.020.
- ^ Miyaura, Norio; Suzuki, Akira (1979). “Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst”. Journal of the Chemical Society, Chemical Communications (19): 866. doi:10.1039/C39790000866.
- ^ Miyaura, Norio; Yamada, Kinji; Suzuki, Akira (January 1979). “A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides”. Tetrahedron Letters 20 (36): 3437–3440. doi:10.1016/S0040-4039(01)95429-2. hdl:2115/44006.
- ^ Nemecek, Gregor; Thomas, Robert; Goesmann, Helmut; Feldmann, Claus; Podlech, Joachim (October 2013). “Structure Elucidation and Total Synthesis of Altenuic Acid III and Studies towards the Total Synthesis of Altenuic Acid II”. European Journal of Organic Chemistry 2013 (28): 6420–6432. doi:10.1002/ejoc.201300879.
- ^ Kürti, László; Czakó, Barbara (2007). Strategic applications of named reactions in organic synthesis : background and detailed mechanisms ; 250 named reactions (Pbk. ed., [Nachdr.]. ed.). Amsterdam [u.a.]: Elsevier Academic Press. pp. 464–465. ISBN 978-0-12-429785-2
- ^ Quach, Tan D.; Batey, Robert A. (April 2003). “Copper(II)-Catalyzed Ether Synthesis from Aliphatic Alcohols and Potassium Organotrifluoroborate Salts”. Organic Letters 5 (8): 1381–1384. doi:10.1021/ol034454n. PMID 12688764.
- ^ Quach, Tan D.; Batey, Robert A. (1 November 2003). “Ligand- and Base-Free Copper(II)-Catalyzed C−N Bond Formation: Cross-Coupling Reactions of Organoboron Compounds with Aliphatic Amines and Anilines”. Organic Letters 5 (23): 4397–4400. doi:10.1021/ol035681s. PMID 14602009.